

Abstract
BACKGROUND: 3-hydroxy-3-methyl-glutaryl coenzyme A (HMG-CoA) reductase inhibitors have several pleiotropic effects, including anti-inflammatory properties, and are reported to improve endothelial functions. Pathophysiologically, acute lung injury (ALI) is caused by a severe inflammatory response and endothelial dysfunction.
OBJECTIVE: To investigate the effects of simvastatin (an HMG-CoA reductase inhibitor) on oxidative stress and lung histopathology in 2 murine models of ALI, induced by oleic acid and endotoxin.
METHODS: The mice were randomly divided into 2 groups: one received 2 mg/kg/d intraperitoneal simvastatin for 15 days. Then the groups were further divided into 3, which received saline, oleic acid, or endotoxin. Four hours after inducing ALI we obtained lung samples for histopathology analysis, myeloperoxidase, glutathione, and malondialdehyde measurement, and blood samples for malondialdehyde measurement.
RESULTS: Endotoxin and oleic acid lung injury increased tissue myeloperoxidase (P = .009 for both), decreased tissue glutathione (P = .02 and P = .009, respectively), and increased tissue malondialdehyde (P = .009 for both), compared to the control group. Simvastatin decreased myeloperoxidase only in the oleic acid group (P = .01). Simvastatin increased glutathione (P = .005 and P = .003, respectively) and lowered malondialdehyde in both the endotoxin and oleic acid groups (P = .003 for both). Histopathology revealed that simvastatin protected the lung tissue in both ALI models, but the protection was greater in the endotoxin group.
CONCLUSIONS: Pretreatment with simvastatin decreased the severity of ALI in oleic acid and endotoxin ALI models, by decreasing inflammation and oxidative stress.
Introduction
Acute respiratory distress syndrome (ARDS) is a common problem, with very high mortality, despite advances in the understanding of its pathogenesis.1 Inflammatory cell activation and increased oxidative stress have been implicated in the pathogenesis.2 No therapies have been shown to be curative, and treatment is mainly supportive, but a lung-protective, low-tidal-volume ventilation strategy increases survival rate by limiting the alveolar damage and consequent biotrauma.3,4
Acute lung injury/ARDS can be caused by pulmonary or extrapulmonary factors, of which by far the most common is sepsis. Pathophysiologically, pulmonary and extrapulmonary ALI/ARDS have some distinct features, despite the common end effects of diffuse alveolar damage and respiratory insufficiency.5 Pathophysiologically, pulmonary ALI is mediated by a direct insult to the alveolar lung cells. The hypoxia is more severe, but the systemic inflammatory response may be limited. Extrapulmonary ALI is mediated by a systemic inflammatory response and reflects the severity of the inflammation. The pulmonary and extrapulmonary forms are reported to have distinct radiological findings and to respond differently to mechanical ventilation strategies. Yet after diffuse alveolar damage and pulmonary edema is established, it is difficult to separate the 2 entities on clinical grounds, and the prognosis is similarly grave for both.
3-hydroxy-3-methyl-glutaryl coenzyme A (HMG-CoA) reductase inhibitors are a class of drugs commonly prescribed for their marked efficacy in reducing blood lipid levels. Soon after the introduction of HMG-CoA reductase inhibitors, it was recognized that they decreased mortality, which could not be explained solely by their lipid lowering effects.6 Subsequent studies revealed that they also have pleiotropic effects that are anti-inflammatory, antioxidant, immunomodulatory, and antithrombotic, and they may improve endothelial dysfunction.6–8
Since, pathophysiologically, ARDS is caused by a severe inflammatory response and is associated with endothelial dysfunction, and in light of recent findings that statins are associated with better outcomes in sepsis and many other inflammatory conditions, we hypothesized that statins might be useful in preventing and treating ARDS.9–11 In an editorial on pandemic influenza, they have also been suggested as a prophylaxis.12 As well, a study by the Irish Critical Care Trials Group reported that statins administered on admission reduced the odds of death in patients with ALI/ARDS.13 However, there have also been contradicting studies that reported that statins did not significantly improve outcomes in patients with pneumonia14 or ALI.15 And those studies had important limitations, which their authors discussed, and this underscores the need to identify statins' mechanisms of action and the need for clinical randomized controlled trials.
We investigated the effects of simvastatin on the development of ALI. We hypothesized that pretreatment with simvastatin would reduce lung injury in both pulmonary and extrapulmonary murine ALI models, by decreasing inflammation and oxidative stress. We induced pulmonary and extrapulmonary ALI with oleic acid and endotoxin, respectively.
Methods
This study was approved by the Institutional Experimental Animal Care and Ethics Committee of Hacettepe University, Ankara, Turkey before the commencement of any intervention (approval number 2006/73–3). The study was performed in the Animal Laboratory of our Department of Pharmacology.
Study Animals
Forty-five Swiss albino mice (weight range 17–53 g) were obtained from our Experimental Animals Breeding Unit, and housed in our Laboratory Animal Husbandry Facility until the experiments. Before and throughout the study, the mice were kept at room temperature (22°C), 30–70% humidity, and 12 hours night/12 hours day conditions. They were fed with standard laboratory food and water ad libitum, and no other restrictions were performed. The mice were assigned to 2 groups. One group received the simvastatin solution, prepared as described below, for 15 days. The control group received daily saline injections (Fig. 1). To minimize first-pass effect of simvastatin, we performed intraperitoneal injection for 15 days, as described below. Five deaths occurred before the intervention, at different time periods, most probably due to the method of activation of simvastatin and consecutive intraperitoneal administrations. 40 mice were included in the study.

Flow chart.
Preparation of the Simvastatin Solution
We obtained the inactive form of simvastatin (Eczacibasi Pharmaceutical, Istanbul, Turkey) and activated it via alkaline hydrolysis, adjusting the pH to 7.2. We administered the simvastatin in intraperitoneal injections at a dose of 2 mg/kg/d in a volume of 10 mL/kg, for 15 days as previously described.16 The control mice received a similar volume of injected saline, with a similar pH.
Induction of Acute Lung Injury
Acute lung injury was induced with oleic acid or endotoxin. The mice in the control group and the simvastatin group were randomly assigned to receive either oleic acid (0.05 mL/kg intravenous to the tail vein), endotoxin (derived from Escherichia coli, lipopolysaccharide O55:B5, 10 mg/kg intraperitoneal, in 0.5 mL), or saline (0.5 mL intraperitoneal). Oleic acid and endotoxin were obtained from Sigma-Aldrich, St. Louis, Missouri. The dose of the endotoxin was chosen on the basis of our previous studies, which found significant in vivo effects at a minimum possible dose.17–19 The timing of endotoxin and oleic acid administration was also chosen on the basis of previous studies, which found that nitric oxide synthase activity reaches its maximum within 2–6 hours after endotoxin administration.20 The mice did not receive supplemental oxygen, fluids, or mechanical ventilation.
Intervention and Sampling
Four hours after inducting acute lung injury, anesthesia was performed with 400 mg/kg chloral hydrate, intraperitoneally. Anesthesia depth was evaluated every 2–3 min throughout the study. The same dose was administered to the different groups, and was found adequate. After adequate anesthesia, we opened the abdomen with a midline (xiphoid to pubis) laparotomy incision, then performed sternotomy and intracardiac blood sampling. The samples were frozen at −80°C, for malondialdehyde measurement. Then the animals were exsanguinated by severing the common carotid artery. One of the lungs was put in 10% formalin solution for histopathologic evaluation, and the other was frozen at −80°C for myeloperoxidase, glutathione, and malondialdehyde measurement.
Myeloperoxidase Assay
Myeloperoxidase was determined as an index of neutrophil accumulation. Tissues were homogenized in 20 mM potassium phosphate buffer (pH 7.4) and the homogenate was centrifuged for 5 min at 10,000 g, at 4°C. The supernatant was discarded, and the pellet was resuspended in 50 mM potassium phosphate buffer (pH 6.0) containing 0.5% hexadecyltrimethylammonium bromide (HETAB). The suspension was frozen and sonicated for 10 seconds. Following incubation for 2 hours in a water bath at 60°C, the suspension was centrifuged at 10,000 g for 5 min. The supernatants were subjected to myeloperoxidase assay, and myeloperoxidase activity was assessed by measuring H2O2-dependent oxidation of o-dianisidin. One unit of enzyme activity was defined as the amount of myeloperoxidase present that caused a change in the absorbance of 1.0/min at 410 nm and 37°C. With the tissue samples we calculated enzyme activity as an indicator of inflammation, in units per milligram of protein.21
Glutathione Assay
We assayed reduced glutathione as an indicator of antioxidant capacity and antioxidant reserves in tissues. We homogenized the tissues (100 mg/mL) in 5% sulfosalicylic acid, and centrifuged the homogenates at 10,000 g for 20 min, then combined 20 μL of the clear supernatant with 160 μL of 0.3 M Na2HPO4 and 20 μL of 0.04% 5,5′-dithiobis-[2-nitrobenzoic acid] in 1% sodium citrate. After 10 min of incubation at room temperature, A405 was read in a microplate reader (Spectramax, Molecular Devices, Sunnyvale, California). The concentration of glutathione was calculated with a standard curve constructed with known concentrations of glutathione, and expressed as μg per 100 mg of wet tissue.22
Malondialdehyde Assay
Plasma malondialdehyde is a marker of systemic inflammation and oxidative stress. We assayed lung tissue malondialdehyde with the thiobarbituric acid assay method of Mihara and Uchiyama.23 We added 3 mL of 1% phosphoric acid and 1 mL of 0.67% thiobarbituric acid solution to a 0.5 mL tissue sample, then heated the mixture in boiling water for 1 hour. After cooling, we added 4 mL of 1-butanol, mixed vigorously, and centrifuged. We then removed the butanol phase and measured absorbance at 532 nm to calculate the malondialdehyde concentration. The malondialdehyde standard was prepared from 1,1,3,3-tetramethoxypropane. We express the plasma and tissue homogenate malondialdehyde concentrations in relation to the protein concentration, which we measured with the method of Lowry et al.24 The results are expressed as nmol per mg of protein.
Histopathology Scoring
After the lung samples were fixed in 10% buffered formalin, they were processed according to routine light microscopy tissue examination methods. A blinded histologist (PA) photographed (DM6000, Leica Microsystems, Buffalo Grove, Illinois) and analyzed 5-μm tissue sections stained with hematoxylin-eosin. We modified scoring systems used in other studies25,26 to histopathologically evaluate neutrophil infiltration and epithelial injury. We used a 0–4 scoring system in which 0 = no infiltration/injury, 1 = minimal infiltration/injury, 2 = mild infiltration/injury, 3 = moderate infiltration/injury, and 4 = severe infiltration/injury.
Statistical Analyses
The myeloperoxidase, glutathione, and malondialdehyde levels and histopathology scores were continuous non-parametric variables, and the results are reported as median and interquartile range. We used the Mann-Whitney U test to compare these continuous non-parametric variables. P < .05 was considered statistically significant. The analyses were conducted with statistics software (SPSS 10.0, SPSS, Chicago, Illinois).
Results
Myeloperoxidase
Figure 2 shows the myeloperoxidase results. The myeloperoxidase levels were higher in the endotoxin and oleic acid groups, as compared to the control group (8.3 [6.7–9.7] U/mg vs 19.2 [17.6–22.3] U/mg, and 8.3 [6.7–9.7] vs 23.2 [19.0–24.3] U/mg, respectively, P = .009 for both). Pretreatment with simvastatin did not significantly change the myeloperoxidase level in the endotoxin group (19.2 [17.6–22.3] U/mg vs 17.3 [13.6–21.7] U/mg, P = .38), but it did decrease the myeloperoxidase level in the oleic acid group (23.2 [19.0–24.3] U/mg vs 15.3 [13.0–18.3] U/mg, P = .01). Simvastatin alone did not change the myeloperoxidase level (8.3 [6.7–9.7] U/mg vs 8.6 [8.4–9.0] U/mg, P = .68).
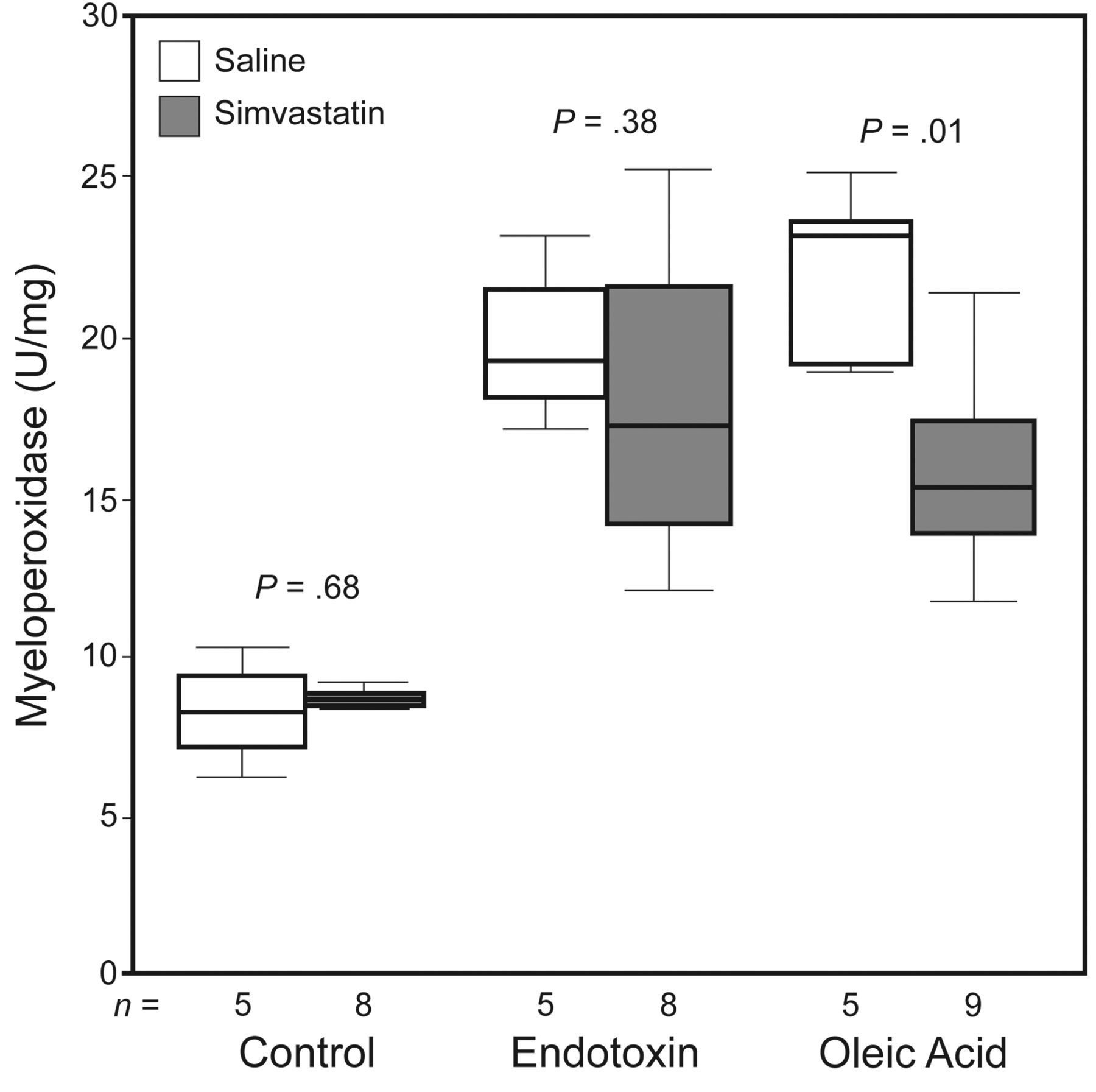
Myeloperoxidase was significantly higher in the mice with acute lung injury/acute respiratory distress syndrome induced with endotoxin or oleic acid (P = .009 for both). In the oleic acid group, the mice pretreated with simvastatin had significantly lower myeloperoxidase. The horizontal lines within the data bars represent the median values. The top and bottom lines of the data bars represent the interquartile range. The whisker bars represent the 1.5 × the upper or lower IQR values.
Glutathione
Figure 3 shows the glutathione results. Compared to the control group, the glutathione levels decreased after endotoxin and oleic acid injection (93.0 [78.0–97.0] μg/100 mg vs 42.0 [29.5–60.5] μg/100 mg, P = .02, and 93.0 [78.0–97.0] μg/100 mg vs 36.0 [31.5–42.0] μg/100 mg, P = .009, respectively). Pretreatment with simvastatin increased glutathione level in both the endotoxin and oleic acid groups (42.0 [29.5–60.5] μg/100 mg vs 72.5 [69.5–86.5] μg/100 mg, P = .005, and 36.0 [31.5–42.0] μg/100 mg vs 60.0 [58.0–63.0] μg/100 mg, P = .003, respectively). Simvastatin alone did not change the glutathione level (93.0 [78.0–97.0] μg/100 mg vs 94.0 [82.0–100.0] μg/100 mg, P = .75).

Glutathione was significantly lower in the mice with acute lung injury/acute respiratory distress syndrome induced with endotoxin or oleic acid (P = .02 and P = .009, respectively). Pretreatment with simvastatin, resulted in higher glutathione in both the endotoxin and oleic acid groups (P = .005 and P = .003, respectively). The horizontal lines within the data bars represent the median values. The top and bottom lines of the data bars represent the interquartile range. The whisker bars represent the 1.5 × the upper or lower IQR values.
Malondialdehyde
Table 1 shows the malondialdehyde results. Compared to the control group, there was no significant difference in plasma malondialdehyde in the endotoxin or oleic acid group (P = .05 and P = .13, respectively). Pretreatment with simvastatin decreased plasma malondialdehyde in the endotoxin group (P = .03), but did not change plasma malondialdehyde in the oleic acid group (P = .88). Simvastatin alone did not change plasma malondialdehyde (P = .66).
Malondialdehyde Results*
Tissue malondialdehyde levels increased after endotoxin and oleic acid injection, as compared to the control group (P = .009 for both). Pretreatment with simvastatin decreased tissue malondialdehyde in both the endotoxin and oleic acid groups (P = .003 for both). Simvastatin alone did not alter tissue malondialdehyde (P = .25).
Histopathology
Table 2 shows the histopathology results. Both endotoxin and oleic acid induced severe neutrophil infiltration and epithelial injury. Pretreatment with simvastatin decreased both neutrophil infiltration and epithelial injury in the endotoxin group (P = .003 and P = .005, respectively), and decreased neutrophil infiltration in the oleic acid group (P = .001).
Histopathology Results*
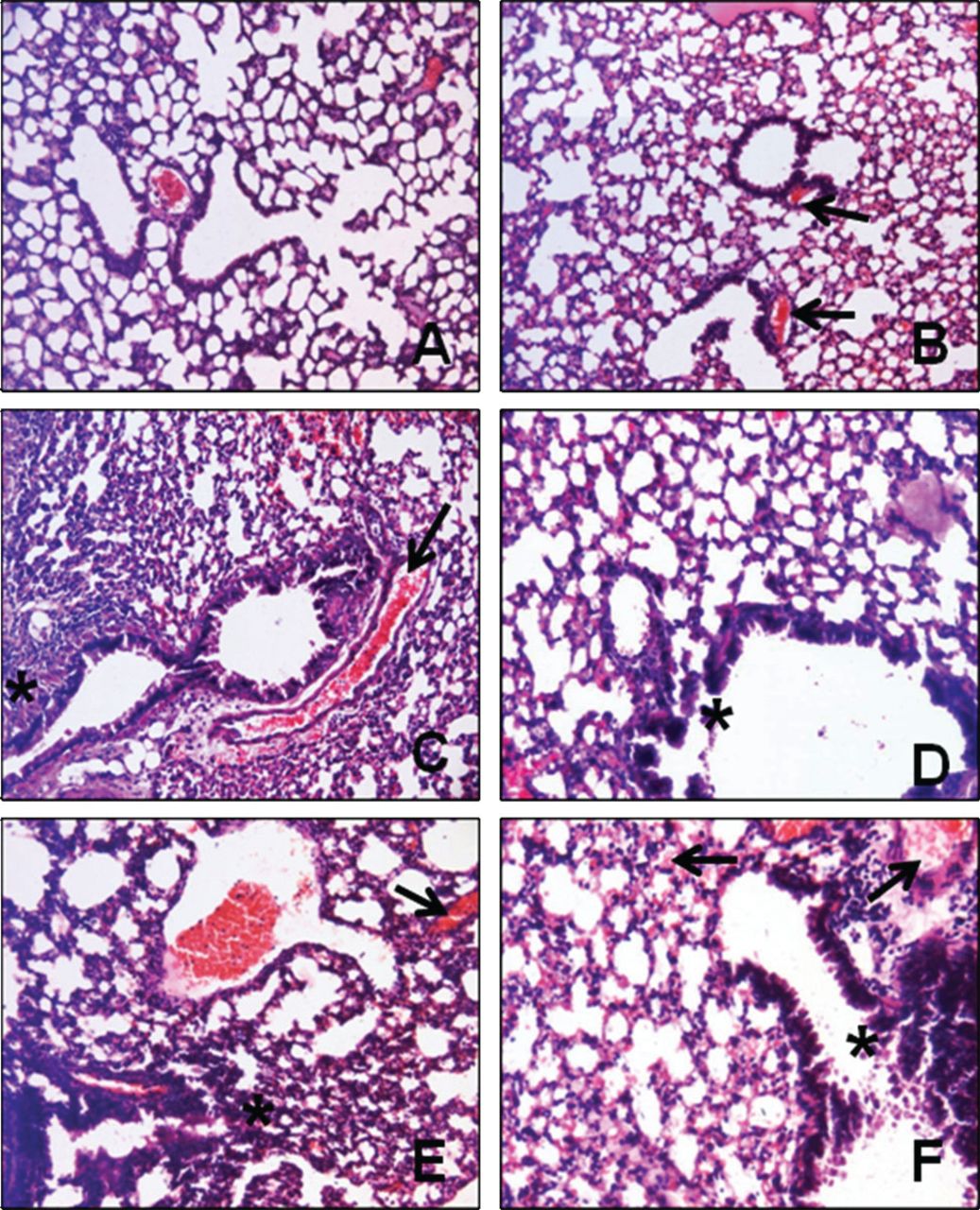
In the control group, normal lung architecture was preserved (Fig. 4A). In the simvastatin without ALI group, all the specimens were histologically almost normal, except for dilatation and stasis of the blood vessels (see Fig. 4B). Similar to the control group, only one of the samples showed mild interalveolar infiltration. Endotoxin damaged both the alveoli and the bronchi (see Fig. 4C). Most of the alveoli were atelectatic, and some of the alveoli and alveolar sacs showed compensatory dilatation. Bronchial epithelium was desquamated. In all of the samples, interalveolar septum was thickened due to infiltration. Most of the samples showed blood vessel dilatation and stasis and interalveolar hemorrhage. Simvastatin protected the alveoli and bronchi from endotoxin (see Fig. 4D); since the alveoli were not collapsed, there was no interalveolar infiltration or compensatory dilatation in the alveoli or alveolar sacs. Four of the samples showed dilatation and stasis only in the capillaries, and 2 samples had dilatation and stasis in all the blood vessels.

A: The control group shows normal lung architecture. B: The simvastatin-only group shows blood vessel dilatation (arrows). C and E: The oleic acid and endotoxin groups show collapsed alveoli (*) and dilatation and stasis of blood vessels (arrow). D: The simvastatin + endotoxin group has less damage than the endotoxin group, but the bronchial epithelium is partly desquamated (*). F: The simvastatin + oleic acid group has less damage than the oleic acid group, but the bronchial epithelium is desquamated (*) and there is substantial blood vessel stasis (arrow). (Hematoxylin-eosin stain, magnification ×200.)
Oleic acid caused alveolar and bronchial damage similar to that caused by endotoxin (see Fig. 4E). Alveolar collapse was very prominent, especially in the peribronchial alveoli. Interalveolar infiltration and thickening of the interalveolar septum due to infiltration were obvious. Simvastatin partially protected against oleic acid injury (see Fig. 4F). There were atelectatic alveoli and compensatory dilatation of the alveoli and alveolar sacs in only in a few areas. Patchy desquamation of the bronchial epithelium was significantly less prominent than in the oleic acid without simvastatin group, but more severe than in the endotoxin with simvastatin group (see Fig. 4D).
Discussion
Pretreatment with simvastatin protected the lung tissue in both ALI models. The protection was more prominent in the endotoxin group, in which there was less epithelial damage and neutrophilic infiltration. We believe the protective mechanism was that simvastatin reduced the oxidative stress from the oleic acid and endotoxin injury. Mice that received simvastatin had lower tissue myeloperoxidase and malondialdehyde levels, higher glutathione levels, and better histopathology scores than mice that did not receive simvastatin. These findings are promising for future clinical studies of HMG-CoA reductase inhibitors as prophylactic agents for ARDS prevention.
The enthusiasm generated by numerous studies revealing positive pleiotropic effects of HMG-CoA reductase inhibitors has stimulated a search for other clinical applications.6 Benefits from HMG-CoA reductase inhibitors have been reported after coronary artery bypass graft, stroke, acute myocardial infarction, revascularization, radiotherapy, and preventing diabetic lung injury in diabetic mice.27–30 Most interestingly, reports indicate potential benefits in sepsis prevention and treatment.9–11 Similarly, there are numerous articles on organ protective effects during sepsis.31–33 There are registered randomized controlled clinical trials underway on statins in sepsis and ALI/ARDS (see http://www.clinicaltrials.gov/ct2/results?term=sepsis+AND+statins).34
However, the available studies on HMG-CoA reductase inhibitors in ALI/ARDS are limited. One study found that simvastatin administered 24 hours before ALI/ARDS induction significantly reduced inflammation variables, vascular leak, and edema formation.35 Pirat et al25 studied lung histopathology, interleukin 1, interleukin 6, tumor necrosis factor alpha, and P-selectin levels in serum and bronchoalveolar lavage samples, and lung tissue malondialdehyde levels, in an intestinal ischemia-reperfusion rat model. They concluded that the protective effects of simvastatin are due to decreased free oxygen radical formation and reduced lung infiltration by leukocytes. Yao et al32 have found reduced vascular leak and inflammation in a different lung injury model. In the light of these findings, Fedson12 suggested HMG-CoA reductase inhibitors as prophylaxis for influenza, because the stock of vaccines and antiviral agents were limited.
We used endotoxin to model extrapulmonary ALI/ARDS, and oleic acid to model pulmonary ALI/ARDS. Endotoxin causes an explosive cytokine release and severe inflammatory response, commonly more severe than what would be normally expected,36 and endotoxin is accepted as a model of extrapulmonary ALI/ARDS.37 It can cause systemic effects and multiorgan dysfunction mediated by neutrophil activation.38 Oleic acid causes marked histopathologic damage similar to ALI/ARDS, independent of neutrophilic infiltration. Oleic acid has minimal systemic effects and is commonly used to model pulmonary ALI/ARDS.39,40 Previous similar studies used shorter duration and higher dosages of simvastatin.25,35 We chose lower simvastatin dosages and prolonged administration to simulate commonly employed clinical simvastatin dosages.
Biochemical Variables
Despite some unexplained aspects of ALI/ARDS pathophysiology, it is commonly accepted that inflammation and oxidative stress have an important role.2 Myeloperoxidase is a marker of neutrophil activation, and glutathione and malondialdehyde are indicators of oxidative stress.25,26,41,42 The myeloperoxidase increase after ALI induction was significantly lower in the groups that received simvastatin. The lack of significant difference in the endotoxin group can only be explained by the fact that endotoxin caused an explosive inflammatory response that could not be totally controlled with simvastatin alone.
Reduction in glutathione level was significantly less after oleic acid or endotoxin in the groups that received simvastatin. As well, the increases in tissue malondialdehyde after ALI induction were less in the simvastatin groups, which indicates lower oxidative load and less lipid peroxidation. Plasma malondialdehyde was increased only in the endotoxin-only group—a finding compatible with endotoxin's ability to cause severe inflammation and multi-organ dysfunction—and pretreatment with simvastatin decreased that response. That we did not find a difference in plasma malondialdehyde in the simvastatin plus oleic acid group could be because oleic acid did not cause a systemic response and the measurements were done 4 hours after ALI induction—a time period that might not be sufficient to detect significant changes in plasma malondialdehyde.
Histopathology Analysis
The protective effect of pretreatment with simvastatin on lung histology tended to be more prominent in the endotoxin group than the oleic acid group. This histopathologic difference, which was not that prominent in biochemical variables, might be due to a greater protective action of simvastatin in generalized inflammation (ie, sepsis) rather than a local effect in pulmonary ALI.9,31–33 One trial reported that HMG-CoA reductase inhibitors themselves did not cause any significant histopathologic changes.35 Yet our finding of vascular dilatation and stasis in the control group that received simvastatin alone deserves further attention for clinical importance. It might reflect increased blood flow with HMG-CoA reductase inhibitors.43 This finding and the protective effects of simvastatin from ALI/ARDS in this study could be due to increased endothelial nitric oxide synthase (eNOS) expression.44 Nitrogen oxide (NO) species are markers of oxidative stress, and are modulated by HMG-CoA reductase inhibitors.45 Endotoxin and inflammatory cytokines activate inducible NO synthase and prolong NO production, which causes hypotension, decreased vasopressor response, and a tendency for multi-organ failure in sepsis, whereas they decrease eNOS.46,47 A recent, multicenter study by McClintock et al45 found that higher urine NO level was associated with better survival, and they suggested that higher NO level is associated with less severe pulmonary and systemic endothelial injury, as indicated by more ventilator-free days and organ-failure free days. Accordingly, statins might benefit ALI/ARDS prevention by increasing eNOS expression. Although the mechanism is yet unclear, studies have reported that HMG-CoA reductase inhibitors increase eNOS expression and simultaneously improve outcome in sepsis.44
Limitations
First, we used oleic acid to simulate pulmonary ALI/ARDS, because oleic acid is known to cause less tumor necrosis factor alpha production, and previous studies reported that oleic acid's systemic effects are minimal.39,40 However, a different model, such as intratracheal saline instillation, could also be used to study simvastatin's effects on pulmonary ALI/ARDS.
Second, we did not evaluate functional alterations and systemic findings because of technical constraints. Small animal models differ markedly from larger animal models: for instance, with a small animal model it is not easy to follow oxygen saturation or conduct multiple blood gas determinations.
Third, because of the study design, we could not evaluate simvastatin's therapeutic effects on ALI/ARDS or the impact on survival. As well, our mice model may only partly resemble ALI in human patients. Our significance tests revealed very low P values, but the study design included multiple statistical tests, which might have increased the probability of type 1 error. Therefore, clinical trials with larger sample sizes are needed to clarify the role of statins in human patients with ALI.
Conclusions
Pretreatment with simvastatin reduced the severity of pulmonary, and especially extrapulmonary ALI/ARDS induced with oleic acid or endotoxin in mice. The protective effect was at least partly mediated by decreases in neutrophil activation, oxidative stress, and lipid peroxidation, but it is difficult to determine their relative contributions to our findings. Many patients are already receiving statins for other reasons. In the light of these findings, they may be at least partially protected against ALI/ARDS. As well, it is highly likely that simvastatins could have a prophylactic value in critically ill patients with high risk of ALI/ARDS. These hypotheses should be studied in human clinical trials, and high priority should be given to study the possible therapeutic effects of simvastatin in patients with ALI/ARDS. However, the clinical importance of the histopathologically observed venous dilatation and stasis in the simvastatin group should also be evaluated.
Acknowledgments
We thank to Banu Cakir MD PhD for statistical assistance, and Beril Talim MD for pathology consultation.
Footnotes
- Correspondence: Arzu Topeli MD, Medical Intensive Care Unit, Department of Internal Medicine, Faculty of Medicine, Hacettepe University, Ankara, 06100 Turkey. E-mail: atopeli{at}hacettepe.edu.tr.
-
Dr Iskit was partly supported by the Turkish Academy of Sciences Young Scientist Award Program (award EA-TUBA-GEBIP/2001–2-11). This research was partly supported by the Hacettepe University Research Fund grant HUAF 02 02 101 034.
- Copyright © 2011 by Daedalus Enterprises Inc.









{kind=link}
{kind=link}
{kind=link}
{kind=link}