
Abstract
Pulmonary veno-occlusive disease is a rare condition with limited treatment options. The pathological hallmark of the disease is occlusion of pulmonary venules and small veins in the lobular septa. The etiology of the disease remains obscure. We report and discuss an extremely unusual case presenting as massive and intractable hemoptysis, in which pulmonary venous occlusion was attributed to granulomatous venulitis in the absence of other pulmonary or systemic inflammatory abnormalities.
- pulmonary veno-occlusive disease
- granulomatous venulitis
- hemoptysis
- pulmonary hypertension
- acute respiratory failure
- ARDS
Introduction
Pulmonary veno-occlusive disease (PVOD) is a rare and poorly understood cause of pulmonary hypertension, characterized pathologically as occlusion of pulmonary veins and venules through endoluminal scarring.1 Clinical findings of pulmonary hypertension, normal pulmonary capillary occlusion (wedge) pressure, and pulmonary edema form a diagnostic triad, but not all elements may be present.2 Confirmation of the diagnosis requires surgical lung biopsy, but frequently the diagnosis is not uncovered until autopsy.
Granulomatous inflammation precipitating PVOD is vanishingly rare, particularly in the absence of systemic disorders such as sarcoidosis. We describe a fatal case of PVOD associated with granulomatous venulitis complicated by pulmonary artery hypertension, massive and persistent hemoptysis, and ARDS.
Case Report
Clinical Presentation
A 27-year-old male presented to the emergency department reporting 4 days of morning hemoptysis and about 500 mL of hemoptysis on the day of admission. He also reported a dry cough with flecks of blood and mild dyspnea on exertion for about 3 months. He had a history of recurrent idiopathic pulmonary emboli 7 years and 6 years prior, reportedly diagnosed by ventilation-perfusion (V̇/Q̇) scans that were high probability for pulmonary embolism. He had been treated with warfarin, with a plan for lifelong anticoagulation following the second event, but this was discontinued for uncertain reasons. He had been admitted to hospital 6 weeks prior with progressive dyspnea on exertion, productive cough, and increasing hemoptysis. At that time, a chest computed tomography scan demonstrated bilateral pulmonary infiltrates, thickened interlobular septae, and mediastinal and hilar adenopathy, but no pulmonary embolism. A V̇/Q̇ scan was intermediate probability for pulmonary embolism. He was treated for community-acquired pneumonia, anticoagulation therapy was resumed, and he was discharged home.
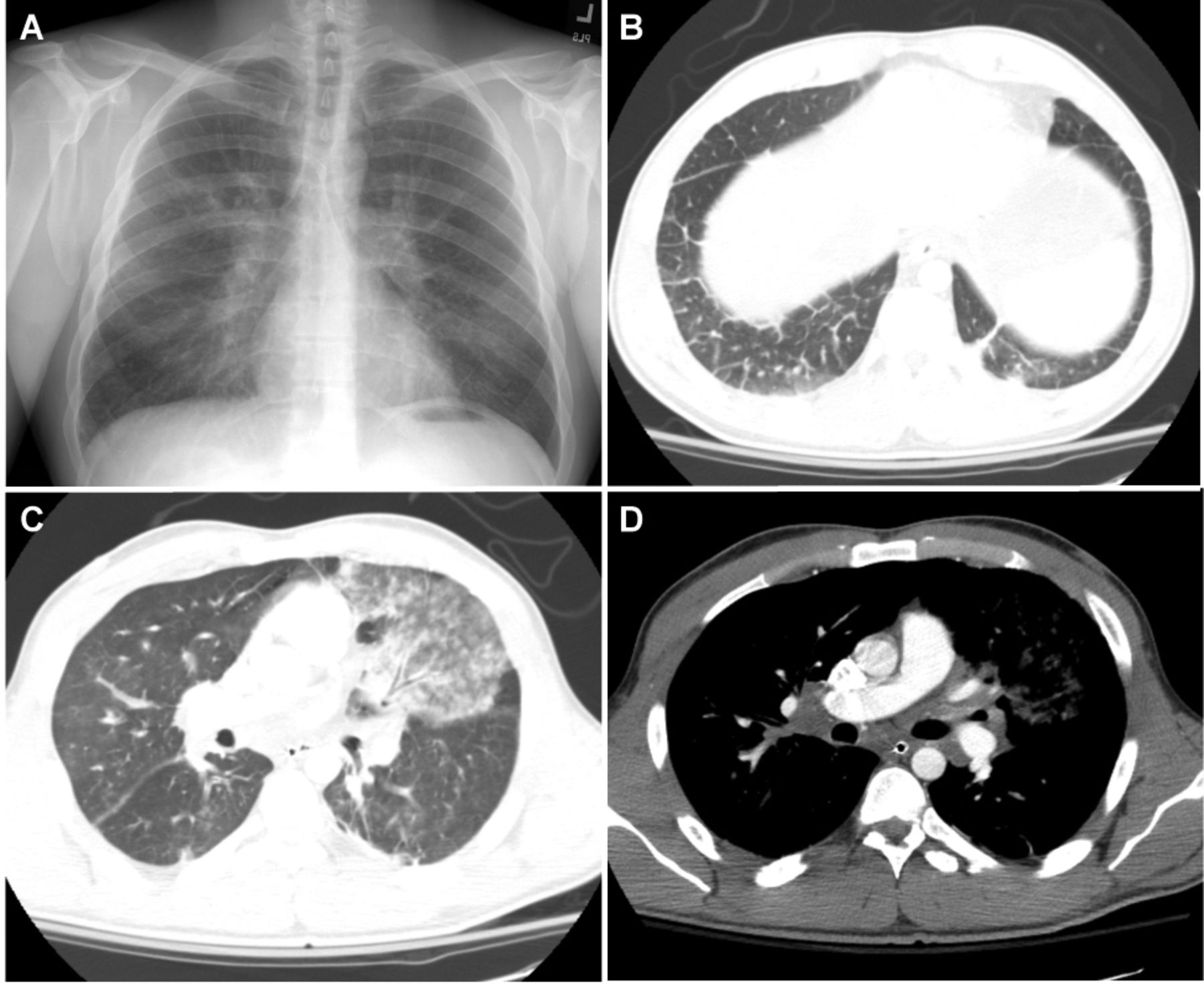
He formerly smoked but had quit 3 months previously. He consumed one alcoholic beverage daily, and had a remote history of marijuana use. He worked as a carpenter. He denied any relevant family history or pertinent review of symptoms. On exam, the patient was tachycardic but normotensive. His breathing frequency was 16 breaths/min, and SpO2 was 96% while breathing ambient air. Sitting upright, he was not in acute distress, but periodically coughed up large quantities of bright red blood. He was unable to tolerate lying supine. Apart from blood in the nares and posterior oropharynx, the exam was unremarkable. Initial laboratory studies showed a white blood cell count of 11.7 × 109/L, hematocrit of 44%, platelets of 286 × 109/L. The international normalized ratio was 2.3, and partial thromboplastin time was 36.6 seconds. Chemistries were all normal. A chest radiograph showed prominent hila bilaterally (Fig. 1) and an electrocardiogram was normal.

A: Admission chest radiograph demonstrating no pulmonary infiltrates but prominent hila. Post-bronchoscopy computed tomogram chest demonstrating (B) thickened interlobular septa and ground glass opacities, (C) parenchymal opacity in the area of bronchoalveolar lavage, and (D) mediastinal and hilar adenopathy.
Urgent bronchoscopy revealed blood in all airways initially, but, with repeated suctioning, slow but persistent bleeding was noted only from the lingular segment. Bronchoalveolar lavage of this segment was stably bloody, confirming ongoing bleeding. A chest computed tomography scan performed immediately thereafter showed an opacity in the lingula, bilateral hilar and mediastinal adenopathy, and coarse septal lines in the lung bases (see Fig. 1). No pulmonary embolism was identified.
The patient's anticoagulated state was reversed, and an inferior vena cava filter was placed. He continued to have substantial hemoptysis requiring several additional bronchoscopies, and embolization of the left bronchial artery. Bleeding persisted and the right bronchial artery was also embolized. Genetic tests subsequently indicated heterozygosity for both the Factor V Leiden mutation and for the prothrombin 20210 mutation. Pulmonary angiography revealed no pulmonary embolism, but pulmonary artery pressures of 77/45 mm Hg. Although he was empirically administered broad spectrum antibiotics, no organisms were cultured from bronchoalveolar lavage samples. On hospital day 3 the patient was brought to the operating room, as the hemoptysis was still not controlled. The left lung was severely congested, and there was a firm area in the lingular segment. Efforts to perform an upper lobectomy were thwarted by bleeding, and a left pneumonectomy was performed instead.
Subsequently, the patient continued to deteriorate. Echocardiography showed severe dilation of the right ventricle and pulmonary artery hypertension, estimated at 68–73 mm Hg. The left ventricle was small, hypercontractile, and had normal systolic function. The patient developed right lung opacities, worsening pulmonary compliance, and became markedly hypoxemic, consistent with ARDS. His hypoxemia was responsive only transiently to inhaled nitric oxide. He developed multisystem organ failure, refractory hypotension and acidemia, and died.
Pathology Examination
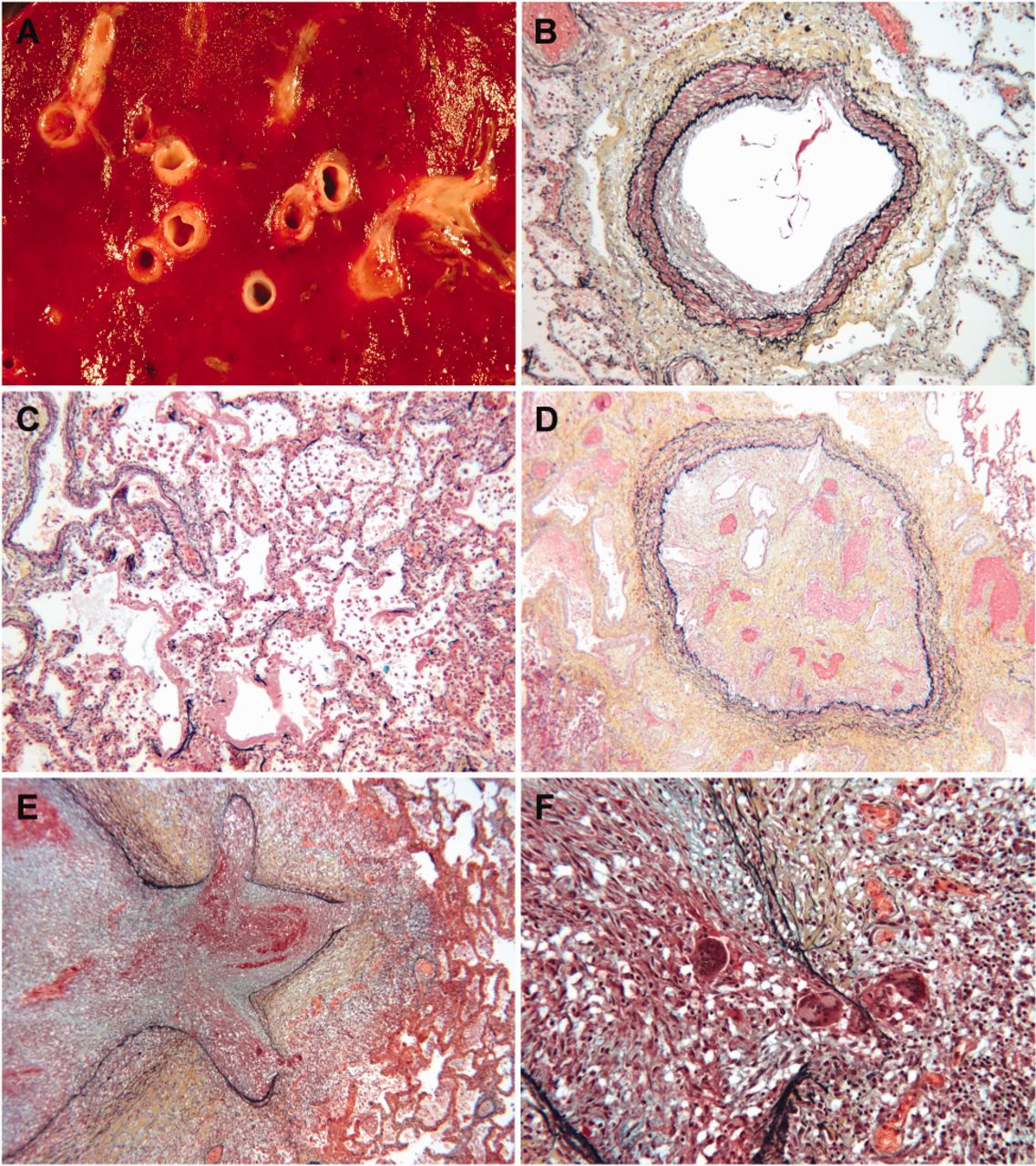
Gross examination of the resected left lung showed congestion and hemorrhage. Microscopic examination identified an occlusive process that preferentially or exclusively involved pulmonary veins. The vascular occlusion was present in several different morphologic stages. Some vessels demonstrated morphological late stage lesions that consisted of complete obliteration of the vascular lumina due to endovascular fibrosis. Morphologically earlier lesions had intense granulomatous inflammation present in the lumina and throughout the walls, with multinucleated giant cells and associated thromboses. Pulmonary arterioles showed mild medial hypertrophy, and larger arteries had intimal plaques, but the airways were normal and no granulomas were found outside occluded veins. There was no evidence of a neoplastic process. Acid-fast and Grocott methenamine silver stains were negative for organisms.
The right lung was examined at autopsy and showed diffuse alveolar damage superimposed on a similar pattern of granulomatous venulitis, pulmonary venous fibrous obliteration, and pulmonary arteriosclerosis (Fig. 2). There was no evidence of recent or remote pulmonary arterial thromboemboli. Acid-fast, periodic acid Schiff, and Grocott methenamine silver stains were negative. Culture of lung tissue grew Candida dubliniensis. There was cardiomegaly with hypertrophy of the right atrium and right ventricle. Mediastinal lymph nodes were uniformly enlarged and had reactive lymphoid follicles and vascular transformation, with only rare giant cells, and no granulomas identified.

{kind=link}
{kind=link}
A: Hemorrhagic right lung with occluded veins. B: Medial hypertrophy of pulmonary artery. C: Hemorrhage and hyaline membrane formation. D: Veno-occlusion with recanalization. Veno-occlusion with granulomatous inflammation at low power (E) and high power (F).
There was no evidence of granulomatous inflammation or vasculitis in other organs, including heart, kidneys, liver, and brain. Post-mortem anti-neutrophil cytoplasmic antibody (ANCA) immunofluorescence was negative and p-ANCA, c-ANCA, and HIV serologies all were negative. The final anatomic diagnosis rendered was PVOD secondary to granulomatous venulitis, with superimposed diffuse alveolar damage.
Discussion
PVOD is a challenging and uncommon diagnosis, typically involving pulmonary venules and veins in lobular septa but sparing larger vessels.3 Tissue diagnosis of PVOD is established by surgical lung biopsy, lung explantation, or, most commonly, at autopsy. Clinically, pulmonary arterial hypertension, a normal pulmonary wedge pressure, and pulmonary edema characterize the disease.2 In this case the diagnosis of PVOD was made pathologically. Clinically, however, the patient had pulmonary hypertension, and the imaging studies were characteristic of PVOD due to the presence of septal thickening, lymphadenopathy, and parenchymal opacities. The fulminant presentation of massive hemoptysis in the setting of anticoagulation for presumed pulmonary emboli complicated the case. Moreover, the pathological feature of isolated granulomatous venulitis was extremely unusual.
The etiology of PVOD is unknown, although infectious insults, including HIV, may be associated with development of disease.4–6 Additionally, toxic exposures, such as to antineoplastic agents, are possible contributors, and PVOD may be associated with bone marrow transplantation.7–12 The association of PVOD with autoimmune disease remains unclear.1 The patient described here had no identifiable risk factors, evidence of infection (the Candida cultured from the right lung was not considered pathogenic), or other clinical or serological features suggesting an occult systemic autoimmune process.
In most reported cases of PVOD, the veins demonstrate fibrous obliteration, with no active injury process. This unusual case of PVOD demonstrates that granulomatous inflammation localized to the pulmonary venule was the underlying etiology, and constitutes only the third such published report.13,14 The etiology of such granulomatous inflammation is unclear. All 3 cases involved young adults (2 males and 1 female) with smoking histories, and the 2 males also reported occasional marijuana use. Others have reported PVOD caused by granulomatous venulitis in the setting of sarcoidosis, a multi-system inflammatory disease often presenting with mediastinal and hilar adenopathy and characterized histologically by the presence of otherwise unexplained noncaseating granulmonas.15–17 In these cases granulomatous inflammation was identified in other tissues.15–17 Furthermore, in an autopsy survey of pulmonary vascular involvement in 40 sarcoidosis cases, 65% of cases had granulomatous venulitis, but none had only vascular involvement.18 However, evidence for pulmonary parenchymal, lymph node, or systemic granulomatous inflammation was absent in this case, arguing against sarcoidosis. The reactive enlargement of the lymph nodes was probably attributable to longstanding lymphatic edema due to post-capillary obstruction in PVOD.
While pulmonary veno-occlusion is the predominant abnormality in PVOD, secondary pulmonary arteriolar thickening is a frequent finding, occurring in 50% of cases. Arteritis and plexiform lesions that may characterize other etiologies of pulmonary arterial hypertension tend to be absent.19 In this case, intimal thickening of pulmonary arteries was apparent on pathological examination and was considered to be secondary to PVOD-induced pulmonary hypertension.
The possibility of thromboembolic disease complicated the diagnosis of PVOD in this case. The patient's heterozygosity for both Factor V Leiden and for the prothrombin 20210 mutations markedly increased his risk of venous thrombosis (by an odds ratio of 20 in one pooled analysis).20 While it was reported that V̇/Q̇ scans 7 and 6 years previously were high probability for pulmonary embolism, no new clinical or pathological evidence obtained in this case was suggestive of acute or chronic pulmonary emboli. Hypoperfusion on a V̇/Q̇ scan has been described as a common finding in PVOD, often leading to an incorrect diagnosis of pulmonary embolism.1 However, a recent study of 56 confirmed or highly probable PVOD cases reported that only 4 (7%) patients had unmatched perfusion defects on V̇/Q̇ scan.21 It therefore seems likely that this patient had pulmonary emboli in the past, but that his more recent symptoms were manifestations of his progressive PVOD and his hemotypsis was worsened by the resumption of anticoagulation therapy.
The massive and intractable hemoptysis that characterized this case of PVOD—and that persisted despite correction of the initial anticoagulated state and repeated therapeutic bronchoscopic and angiographic interventions—is unusual. Because of post-capillary obstruction, alveolar hemorrhage is more common with PVOD than with pre-capillary pulmonary arterial hypertension, but is frequently occult.22 Although the hemorrhage initially appeared to be localized, the dire and extensive interventions necessitated, and the pathological findings indicated, the diffuse nature of the process. The progression to ARDS, manifested pathologically as diffuse alveolar damage, was associated with multisystem organ failure.
Since being recognized in the 1930s,23 treatment of PVOD remains largely unsuccessful. Vasodilators, a mainstay of pulmonary hypertension therapy, may in fact be deleterious by increasing pulmonary arterial flow in the setting of fixed venous obstruction, leading to pulmonary edema. Immunosuppressants and anticoagulation are sometimes used, but with little objective evidence of benefit. In this rare case, corticosteroid therapy to reduce the granulomatous inflammation might have been an effective treatment strategy had the diagnosis been made earlier. Lung transplantation is the most beneficial therapy for PVOD, but patients require prompt referral after diagnosis as survival time is less than 2 years.
Footnotes
- Correspondence: T Eoin West MD MPH, Division of Pulmonary and Critical Care Medicine, Department of Medicine, Harborview Medical Center, University of Washington School of Medicine, 325 9th Avenue, Box 359640, Seattle WA 98104. E-mail: tewest{at}uw.edu.
The authors have disclosed no conflicts of interest.
- Copyright © 2013 by Daedalus Enterprises
References
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.
- 10.
- 11.
- 12.
- 13.
- 14.
- 15.
- 16.
- 17.
- 18.
- 19.
- 20.
- 21.
- 22.
- 23.