
Abstract
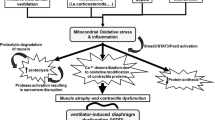
Experimental evidence suggests that controlled mechanical ventilation (CMV) can induce dysfunction of the diaphragm, resulting in an early-onset and progressive decrease in diaphragmatic force-generating capacity, called ventilator-induced diaphragmatic dysfunction (VIDD). The mechanisms of VIDD are not fully elucidated, but include muscle atrophy (resulting from lysosomal, calpain, caspase and proteasome activation), oxidative stress, structural injury (disrupted myofibrils, increased numbers of lipid vacuoles, and abnormally small and disrupted mitochondria), myofiber remodeling and mitochondrial dysfunction.
The major clinical implication of the VIDD is to limit the use of CMV to the extent possible. Partial (assisted) modes of ventilatory support should be used whenever feasible, since these modes attenuate the deleterious effects of mechanical ventilation on respiratory muscles.
Similar content being viewed by others

Introduction
Controlled mechanical ventilation (CMV) is a mode of ventilatory support where the respiratory muscles are not contracting and the ventilator takes full responsibility for inflating the respiratory system. Animal studies suggest that CMV can induce dysfunction of the diaphragm, resulting in decreased force-generating capacity, called ventilator-induced diaphragmatic dysfunction (VIDD) [1].
The frequency of CMV use cannot be determined with certainty. The international mechanical ventilation study group reported that 13% of mechanically ventilated patients receive a neuromuscular blocker for 8% of the total days of ventilatory support [2]. In these patients full ventilatory support is mandatory. There are additional patients who are on full ventilator support without receiving neuromuscular blockers (e.g. traumatic brain injury patients, postoperative neurosurgical patients, comatose patients, and patients with status epilepticus on barbiturate coma to suppress seizure activity). Thus, a significant proportion of mechanically ventilated patients are on full ventilator support and could be potentially vulnerable to VIDD.
The mechanisms of this dysfunction and the clinical relevance for mechanically ventilated patients will be the subject of this review.
Evidence from animal models
Diaphragmatic force and endurance following CMV
Measurements in intact animals
Controlled mechanical ventilation leads to decreased diaphragmatic force-generating capacity in various animal species. In the intact diaphragm studied in vivo, transdiaphragmatic pressure (Pdi) generation upon phrenic nerve stimulation declines at all stimulation frequencies (20–100 Hz) (Fig. ESM 1) [3–6]. The decline ensues early (1 day in rabbits [5], 3 days in piglets [4, 6]) and is progressive, Pdi decreasing to 63% of the control value after 1 day of CMV and to 49% of the control value after 3 days of CMV in rabbits [5]. Within a few days (3 days in rabbits, 3–5 days in piglets, 11 days in baboons) the pressure generating capacity of the diaphragm declines by 35–50%. The endurance of the diaphragm is also compromised [3].
The decreased force-generating capacity ensuing with CMV is not due to changes in lung volume [6] or to changes in abdominal compliance [3, 4]. Neural and neuromuscular transmission remain intact, as evidenced by the lack of changes in phrenic nerve conduction (latency) and the stable response to repetitive stimulation of the phrenic nerve [4] (Fig. 1). In contrast, the compound muscle action potential (CMAP) declines progressively, suggesting that excitation/contraction coupling or membrane depolarization may be involved (Fig. 1) [4]. Thus, the CMV-induced impairment in the diaphragmatic force-generating capacity appears to reside within the myofibers.

The evoked compound muscle action potential (CMAP) tracings of the diaphragm upon electrical stimulation of the phrenic nerves in vivo from one piglet on days 1, 3 and 5 of CMV. The time from the stimulus to the onset of CMAP (Latency) does not change after 3–5 days of CMV, whereas the amplitude of CMAP is progressively reduced. From reference [4] with permission. This indicates that the neural and neuromuscular transmission are not affected when VIDD develops and that the contractile dysfunction resides within the diaphragmatic myofibers
In vitro measurements
The isometric (both twitch and tetanic) tension development by isolated diaphragmatic strips in vitro [7–10] confirm the in vivo findings and suggest that the decline in contractility is an early (12 h) and progressive phenomenon (Fig. 2) [8], the isometric force declining by 30–50% after 1–3 days of CMV in rats. The force–velocity relationship of the diaphragm also changes, the maximum shortening velocity increasing after CMV [11] (Fig. ESM 2; for details and implications see the ESM text).

Effects of prolonged CMV on the diaphragmatic force–frequency response in vitro in rats. Values are means ± SE. Compared with control, CMV (all durations) resulted in a significant (* p < 0.05) reduction in diaphragmatic specific force production at all stimulation frequencies. From reference [8] with permission. Please note that the decline in force is progressive, worsening as the duration of CMV is prolonged
The effects of CMV on diaphragm in vitro fatigability are controversial (see ESM).
Pathophysiology
Muscle atrophy
CMV leads to diaphragmatic atrophy [7, 9, 10, 12]. Ventilator-induced atrophy [13] develops rapidly (as early as 12 h after the institution of CMV [14]) and is more pronounced in the diaphragm, which atrophies earlier than the peripheral skeletal muscles that are also inactive during CMV [7, 9, 12]. Two days of CMV with PEEP (2 cmH2O) induced atrophy in rabbits [10], whereas 3 days of CMV without PEEP were inadequate to induce atrophy [5], which suggests that the rapidity of atrophy development might be augmented with the use of PEEP. The increased lung volume at the end of expiration with the use of PEEP would put the passive diaphragm in a relatively shortened position, and skeletal muscles atrophy faster in the shortened position [15, 16].
The decreased volume of the cytoplasm (atrophy) was observed in the presence of decreased number of myonuclei (skeletal muscle cells are multinucleated cells and theoretically a single myonucleus can sustain the necessary gene expression for a limited area of the cytoplasm, a relationship known as the myonuclear domain [17]), so that the myonuclear domain remains constant [14]. This decrease in myonuclear content was mediated by caspase-3-dependent increased apoptosis, which was evident as early as 6 h after the onset of CMV [14]. Both the apoptosis and the atrophy were attenuated with caspase-3 inhibition [14].
Atrophy can result from decreased protein synthesis, increased proteolysis or both. Six hours of CMV in rats decreased the in vivo rate of mixed muscle protein synthesis (an average synthesis rate for all muscle proteins) by 30% and the rate of myosin heavy chain protein synthesis by 65%, both of which persisted throughout 18 h of CMV [18]. In addition, 24 h of CMV suppressed the mRNA levels of insulin-like growth factor (IGF)-1, which stimulates protein synthesis [19]. Thus, CMV decreases protein synthesis in the diaphragm.
Increased proteolysis has been documented in diaphragm strips of animals subjected to 18 h of CMV [12]. All systems of proteases that mammalian cells have for intracellular proteolysis (the lysosomal proteases, the calpains, the caspases and the proteasome system) [20]), are activated after CMV [12, 14, 21]. Calpains do not fully degrade, but only partially cleave proteins in vivo (Fig. ESM 3). This renders the proteins amenable to the proteasome [13]. The stimulus for calpain activation in not known, but calcium elevation in the cell is prerequisite. The reduced (mRNA) levels of sarcoplasmatic reticulum calcium ATPase (the enzyme that removes calcium from the sarcoplasm) secondary to 24 h of CMV [22] may contribute to calpain activation. The lysosomal proteases such as cathepsin B are also activated secondary to CMV [21]. Caspases are proteases that can degrade proteins and especially complexes of actin and myosin [20, 23]. Upregulation of caspase-3 expression has been documented in the diaphragm secondary to CMV [14]. Caspase-3 can be activated by oxidative stress, increased intracellular calcium and increased calpain activity [20].
Using the proteasome inhibitor lactacystin, Shanely et al. [12] showed that the proteasome is involved in the augmented proteolysis of the diaphragm strips from CMV animals. The proteasome is a multi-subunit multi-catalytic complex that exists in two major forms (Fig. 3): the core 20S proteasome can be free or bound to a pair of 19S regulators to form the 26S proteasome, which degrades (in an ATP-dependent manner) proteins covalently bound to a polyubiquitin protein chain (ubiquitinated proteins). The binding of ubiquitin to protein substrates requires the ubiquitin-activating enzyme (E1), which utilizes ATP-derived energy to form a covalent link with a ubiquitin protein, followed by transfer of the active ubiquitin moiety to a ubiquitin-conjugating enzyme (E2) and finally transfer of this ubiquitin to the protein to be degraded via a ubiquitin ligase (E3). Accordingly, CMV increases the level of ubiquitin–protein conjugates in the diaphragm [24] that are the substrates of the 26S proteasome (Fig. 3) [25]. Furthermore, key enzymes involved in the function of ubiquitin–proteasome pathway are upregulated in the diaphragm, such as the skeletal muscle-specific ubiquitin ligases (E3 enzymes) muscle atrophy F-box (MAFbx/Atrogin-1) [24, 26, 27] and muscle ring finger-1 (MuRF1) [24, 27]. However, not all mRNAs of the ubiquitin–proteasome pathway are upregulated in the diaphragm, since no change was found for the ubiquitin-conjugating enzyme E214k, or the polyubiquitin secondary to 12 h of CMV [24]. The relatively short period of CMV (12 h) may have been inadequate for the upregulation of these components of the ubiquitin–proteasome pathway. In any case, increased protein ubiquitination in the diaphragm secondary to CMV did not require increased expression of all components of the ubiquitin-proteasome pathway.

The ubiquitin–proteasome pathway of proteolysis. Proteins degraded by the ubiquitin–proteasome pathway are first conjugated to ubiquitin (Ub). The process of linking ubiquitin to lysine residues in proteins destined for degradation (inlet) involves the activation of ubiquitin by the E1 enzyme in an ATP-dependent reaction. Activated ubiquitin is transferred to an E2 carrier protein and then to the substrate protein, a reaction catalyzed by an E3 enzyme (E3 ligase). This process is repeated as multiple ubiquitin molecules are added to form a ubiquitin chain. In ATP-dependent reactions, ubiquitin-conjugated proteins are recognized and bound by the 19S complex, which releases the ubiquitin chain and catalyzes the entry of the protein into the 20S core proteasome. Degradation occurs in the 26S core proteasome, which contains multiple proteolytic sites within its two central rings. Peptides produced by the proteasome are released and rapidly degraded to amino acids by peptidases in the cytoplasm or transported to the endoplasmic reticulum and used in the presentation of class I antigens. The ubiquitin is not degraded but is released and reused. SH denotes sulfhydryl, PP 1 pyrophosphate, and ATP, ADP adenosine tri- and diphosphate respectively. From reference [25] with permission
Interestingly, Shanely et al. showed that CMV resulted in 500% increase in the 20S proteasome activity [28], which is specialized in degrading proteins oxidized by reactive oxygen species. Oxidative damage of a protein results in its partial unfolding, exposing hidden hydrophobic residues (Fig. ESM 4) [29, 30]. Therefore, an oxidized protein does not need to be further modified by ubiquitin conjugation to confer a hydrophobic patch, nor does it require energy from ATP hydrolysis to unfold.
Oxidative stress
CMV is associated with augmented oxidative stress in the diaphragm, as indirectly evidenced by the rise in protein oxidation (elevated protein carbonyls [12]; Fig. 4) and lipid peroxidation (elevated 8-isoprostane [12], total lipid hydroperoxides [27, 28] and thiobarbituric reactive substance content [6]) and directly shown by the increased emissions of dichlorofluorescein (a molecule that fluoresces upon reacting with reactive oxygen species within cells) when diaphragmatic strips from CMV-treated animals are incubated in vitro with the dye [31]. The onset of oxidative injury is rapid, occurring within 6 h after the institution of CMV in rats [28], and is long-lasting, being present after 3 days of CMV in piglets [6].

Illustration of Western blots using monoclonal antibodies to identify oxidized proteins with molecular masses of 200–40 kDa. Left lane: Reactive carbonyl derivatives (RCD), which are the footprints of protein modifications induced by oxidative stress in insoluble proteins isolated from the diaphragm of an animal exposed to CMV for 18 h. Middle and right lanes: The same membrane stripped of the 2,4-dinitrophyenylhydrazone antibody (the antibody recognizing RCD was removed) and then sequentially re-probed with monoclonal antibodies specific for rat skeletal muscle actin and all myosin heavy chain (MHC) isoforms. From reference [28] with permission
The response of antioxidant enzymes in the diaphragm to CMV is controversial and the mechanisms of oxidative stress generation remain elusive (for both see ESM).
Oxidative stress can modify proteins involved in energetics, excitation–contraction coupling, intracellular calcium regulation and force generation [32]. CMV-induced diaphragmatic protein oxidation was evident in proteins with molecular masses of about 200, 128, 85, and 40 kDa [28]. These findings raise the possibility that actin (40 kDa) and/or myosin (200 kDa) undergo oxidative modification during CMV (Fig. 4), which would be expected to compromise diaphragm contractility. This intriguing possibility awaits confirmation by more specific identification of the modified proteins.
Oxidative stress and especially the lipid peroxidation product 4-hydroxy-2-nonenal (produced in the diaphragm under conditions such as sepsis or resistive loading [33]) can reduce the activity of plasma membrane calcium ATPase [34]. This would retard calcium removal from the diaphragmatic myofibers and would contribute to calcium accumulation and calpain activation [20]. Oxidative stress could also injure various intracellular structures (organelles).
Structural injury
Structural abnormalities of different subcellular components of diaphragm myofibers progressively develop after 2–3 days of CMV in rabbits (Fig. ESM 5) [5, 11, 35]. The changes consisted of disrupted myofibrils, increased numbers of lipid vacuoles in the sarcoplasm, and abnormally small mitochondria containing focal membrane disruptions. Similar alterations were observed in the external intercostal muscles of ventilated animals [35] but not in the hindlimb muscle [5]. The structural abnormalities have detrimental effects on diaphragmatic contractility. The number of abnormal myofibrils is inversely related to the force output of the diaphragm [5]. The mechanisms of injury have not been elucidated, but may involve activation of calpains, which have the ability to degrade several sarcomeric proteins and direct cellular injury secondary to augmented oxidative stress [1].
Muscle fiber remodeling
Muscle fibers are classified as either slow-twitch (type I) or fast-twitch (type II) based on their myosin heavy chain (MHC) isoform content (in increasing order of maximum shortening velocity, MHC isoforms are I, IIa, IIx and IIb) [36]. Muscles can modify their MHC phenotype in two ways: preferential atrophy/hypertrophy of fibers containing a specific MHC isoform and actual transformation from one fiber type to another. Both short-term [12, 22] and long-term [9] CMV result in significant modifications of the MHC phenotype in rats. Within 12 and 18 h of CMV both type I and II fibers are reduced in size [12, 14], yet type II fibers exhibit much greater reduction [12], and within 24 h of mechanical ventilation the transcript levels of the MHC 2A and 2B isoforms are decreased by ∼20% [22], consistent with the preferential atrophy observed in the above-mentioned studies. This modification of the MHC phenotype could contribute to the force decline of the diaphragm, since the force produced by slow fibers is less than the force produced by fast fibers [37], but at the same time could explain the increased fatigue resistance observed [38], (though this result is not uniform in all studies), since type I fibers have higher oxidative capacity and thus endurance than type II fibers. However, prolonged (44–93 h) CMV in rats results in a different pattern of MHC phenotype modification, with decreased number of type I fibers and increases in fast MHC isoforms mainly within hybrid fibers (fibers co-expressing both slow and fast isoforms) [9]. This slow-to-fast transformation does not compromise diaphragmatic contractility per se, but reduces diaphragmatic endurance, since fewer slow-twitch fatigue-resistant fibers are available. The basis of this different response is not known, but may be related to the ventilatory strategy or the duration of CMV (< 24 h vs. 44–93 h). Despite similar respiratory rates, the tidal volume and thus the degree of phasic diaphragmatic shortening was double in the study of Shanely et al. [12] (1 ml/100 g body weight) compared to the tidal volume used by Yang et al. [9] (0.5 ml/100 g body weight). In contrast, the degree of tonic diaphragmatic shortening imposed by positive end-expiratory pressure (PEEP) was much greater in the study of Yang et al. [9] (PEEP = 4 cmH2O) than in the study of Shanely et al. [12] (PEEP = 1 cmH2O). Whether different ventilatory patterns result in different fiber type transformations is not known. Interestingly, in limb muscles the duration of inactivity influences the fiber type transformation observed. Whereas short-term inactivity results in fast-to-slow transformation (similar to short-term mechanical ventilation), longer inactivity results in slow-to-fast transformation. Although the duration of inactivity was much longer (6 weeks) in limb muscles, the diaphragm might exhibit much faster adaptation to inactivity, since it is continuously contracting throughout life with much higher duty cycles (duration of contraction relative to relaxation) than the limb muscles. In rabbits, 2 days of CMV resulted in atrophy of the respiratory muscles and in decreased cross-sectional area of type IIa and IIb fibers but not type I fibers (fast-to-slow transformation), with no change in their proportion [10], whereas 3 days of mechanical ventilation did not affect the cross-sectional areas of slow and fast fibers but resulted in a decrease in the proportion of the fast MHC2X isoform (a form of fast-to-slow transformation) [5]. The differences might be attributed to the presence of PEEP (2 cmH2O in [10], 0 cmH2O in [5]) or to the episodes of breakthrough diaphragmatic activity observed [5].
Interestingly, 24 h of CMV in rats resulted in changes in the mRNA expression of the myogenic regulatory transcription factors myoD (myogenic determination gene D) (decrease) and myogenin (increase) with a consequent decrease in the myoD/myogenin ratio [22]. These transcription factors bind to the promoter of many skeletal muscle-specific genes and drive myoblast determination and differentiation during embryogenesis, but may also influence fiber type transformation in the adult muscle. The levels of myoD are significantly greater in fast than in slow muscle fibers, myogenin is preferentially located in slow fibers, and their ratio is highly correlated with muscle fiber phenotype. Whether this correlation is causal is debated [39]. In the diaphragm, the deletion of MyoD resulted in a shift in MHC phenotype from MHC IIb toward the slower MHC IIa and IIx (fast to slow transformation), associated with decreased force-generating capacity [40]. Thus, the decreased myoD/myogenin ratio in the diaphragm secondary to 24 h of CMV [22] might contribute to the observed fast-to-slow fiber type transformation.
Remodeling could also lead to reduced optimal diaphragmatic length [7, 9] (see ESM).
Drugs
Anesthetics should be excluded as causes of VIDD, since studies that used appropriate controls (to the extent feasible, i.e. a group of anesthetized spontaneously breathing animals) concluded that the decreased contractility was due to the effects of mechanical ventilation per se (and not to anesthetic use) [5].
Neuromuscular blockers [3, 9] cannot solely account for the decreased contractility secondary to CMV, since decreased contractility was also observed in studies that did not use these drugs [5, 8]. However, the effects of 24 h of CMV and aminosteroidal neuromuscular blockers (rocuronium) are synergistic in depressing diaphragm contractility, in inducing atrophy of type IIx/b fibers and in upregulating the ubiquitin ligase (E3) MuRF1 (but not the E3 MAFbx/Atrogin-1) [27], a synergism not observed with different doses of benzylisoquinoline neuromuscular blockers (cisatracurium) [41].
Metabolic enzymes and mitochondrial function
The changes documented after CMV are not dramatic (see ESM).
Clinical relevance
Evidence for VIDD in humans
Although there is no definite evidence of VIDD in humans, several intriguing data suggest that it is a clinical relevant phenomenon. The twitch transdiaphragmatic pressure elicited by magnetic stimulation of the phrenic nerves was reduced in mechanically ventilated patients [42] and in patients ready to undergo weaning trials [43] compared to normal subjects. This is not specific evidence for the presence of VIDD, since other factors leading to muscle weakness in the ICU may have contributed. Diaphragmatic atrophy was documented (by ultrasound) in a tetraplegic patient after prolonged CMV [44]. However, denervation removes neurotrophic influences for the muscle, which is not the case for VIDD. Retrospective analysis of postmortem data obtained in neonates who received ventilatory assistance for 12 days or more immediately before death documented diffuse diaphragmatic myofiber atrophy (small myofibers with rounded outlines), not present in extradiaphragmatic muscles [45]. Furthermore, preliminary data suggest that brain-dead organ donors (with an intact circulation) who underwent CMV for 18–72 h exhibit reduced cross-sectional area (i.e. atrophy) of both slow and fast diaphragmatic fibers (by 40% and 36% respectively) compared to matched control patients subjected to surgery for resection of solitary pulmonary nodules (receiving CMV for less than 2 h) [46]. The ubiquitin–proteasome pathway was implicated in the development of atrophy, since both the ubiquitin–protein conjugates and the mRNA levels of the E3 ligases MAFbx/Atrogin-1 and MuRF1 were upregulated in the diaphragms of brain-dead patients receiving CMV [47].
Clinical context
VIDD should be suspected in patients who fail to wean after a period of CMV. The weaning failure is related to respiratory muscle weakness. Other causes of respiratory muscle weakness should be ruled out [48]. However, the above-mentioned conditions may coexist with VIDD.
VIDD prevention
Ventilatory strategy
Since data in humans are lacking, suggestions are based on animal models and speculations. The time spent in CMV must be curtailed to the extent possible, especially in older individuals, since the effects of aging and CMV are additive [49]. Although CMV induced similar losses (24%) in diaphragmatic isometric tension in both young and old animals, the combined effects of aging and CMV resulted in a 34% decrement in diaphragmatic isometric tension compared to young control animals.
When feasible, partial support modes should be used. Recent studies raise the possibility of partial support modes in conditions traditionally considered as indications for CMV such as ALI/ARDS [50, 51]. In an animal model, assisted (flow-triggered pressure-limited) mechanical ventilation from the onset of ventilator support resulted in attenuation of the force loss induced by CMV (Fig. 5) [26]. Thus, it stands to reason that preserving diaphragmatic contractions during mechanical ventilation should attenuate the force loss induced by CMV, though other forms of partial ventilatory support (pressure support, SIMV) have not been experimentally tested. It should be stressed that these suggestions may be valid in the absence of sepsis, since during sepsis in rats (albeit of short duration, 4 h), CMV protects the diaphragm from injury [52].

Diaphragmatic tetanic force at various stimulation frequencies in control circumstances, assisted mechanical ventilation (AMV), and controlled mechanical ventilation (CMV) in rats. Values are mean ± SE. * p < 0.01, CMV versus control and AMV. CSA, cross-sectional area. From reference [26] with permission
Assisted modes or even noninvasive mechanical ventilation in hypercapnic COPD [53–55] can be an alternative strategy in patients who experience weaning failure after a spontaneous breathing trial or after extubation and who may be ventilated using CMV [56], a strategy based on the premise that respiratory muscle fatigue (requiring rest to recover) is the cause of weaning failure [57, 58]. This is because the load that the respiratory muscles of patients who fail to wean are facing is increased to the range that would predictably produce fatigue of the respiratory muscles [59], if patients were allowed to continue spontaneous breathing without ventilator assistance. Recent evidence, however, does not support the existence of low-frequency fatigue (the type of fatigue that is long-lasting, taking more than 24 h to recover) in patients who fail to wean despite the excessive respiratory muscle load [43]. This is because physicians have adopted criteria of spontaneous breathing trial failure and termination of unassisted breathing, which lead them to put patients back on the ventilator before the development of low-frequency respiratory muscle fatigue. Thus, no reason exists to completely unload the respiratory muscles with CMV for fatigue reversal if weaning is terminated based on predefined criteria [56].
Intermittent diaphragmatic contractions
When CMV is inevitable, short periods of diaphragmatic activity have been suggested as a preventive countermeasure. This could be achieved with either phrenic nerve stimulation or short periods of intermittent spontaneous breathing. Only 30 min of pacing of one hemidiaphragm each day attenuated atrophy in this hemidiaphragm during prolonged CMV in a tetraplegic patient compared to the non-paced hemidiaphragm [44]. In rats subjected to 24 h of CMV, either 5 min or 60 min of spontaneous breathing every 6 h did not preserve diaphragm force. Rats receiving CMV developed reduced cross-sectional areas of type I and type IIx/b diaphragmatic fibers, which was not observed in intermittently spontaneously breathing rats, yet no difference was observed in the cross-sectional areas between the CMV rats and the intermittently spontaneously breathing rats [60]. Whether more frequent or longer intervals of spontaneous breathing might be more effective in preventing VIDD awaits experimental proof.
Pharmacological approaches
Antioxidant supplementation could decrease the oxidative stress and thus could attenuate VIDD. Accordingly, when rats were administered the antioxidant Trolox (an analogue of vitamin E) from the onset of CMV, its detrimental effects on contractility (Fig. 6) and proteolysis were prevented [61].

Force–frequency curves of in vitro diaphragm strips from control rats (CON), spontaneously breathing animals (SBS), mechanically ventilated animals (MVS), and mechanically ventilated animals receiving Trolox (MVT). Values represent means ± SEM. * Significantly different from CON group, p < 0.05; + significantly different from SBS group, p < 0.05; # significantly different from MVT group, p < 0.05. From reference [61] with permission
A similar approach is adopted by nature itself! Various dormant animals immobilized for prolonged periods of time prevent muscle atrophy through a decrease in metabolic rate that reduces formation of reactive oxygen species and through a concomitant rise in antioxidant enzymes [62, 63]. Interestingly, a combination of vitamins E and C administered to critically ill surgical (mostly trauma) patients was effective in reducing the duration of mechanical ventilation compared to nonsupplemented patients [64]. It is tempting to speculate that part of this beneficial effect was mediated by preventing VIDD. Thus, when CMV is used, concurrent administration of antioxidants seems justified, since a recent meta-analysis suggests that they are beneficial in critical care patients [65].
Administration of leupeptin (an inhibitor of lysosomal proteases and calpain) at the beginning of CMV prevented the development of diaphragmatic contractile dysfunction and atrophy [21] in experimental animals. This raises the possibility of future clinical trials of protease inhibitors in patients to prevent VIDD.
Recovery from VIDD
There is no established or experimentally tested therapy for VIDD. Theoretically, resumption of spontaneous breathing would retrain the respiratory muscles, yet the time course of recovery of normal function is unknown. A major concern is that diaphragm disuse associated with CMV would increase its susceptibility to subsequent contraction-induced injury, once respiratory efforts are resumed, similar to other skeletal muscles [66]. Rats receiving 24 h of CMV exhibited 26% decline in maximal specific diaphragmatic force with no apparent injury to the cell membrane or evidence of inflammation [67]. Resumption of spontaneous breathing for 2 h in these rats did not exacerbate contractile dysfunction or induce membrane injury or macrophage invasion [67]. However, reloading was associated with increased myeloperoxidase activity and neutrophil infiltration in the diaphragm, which is expected to cause injury at a later time point, should reloading be continued [67]. Further studies are needed to elucidate the recovery response of the diaphragm that has developed VIDD to the resumption of spontaneous respiratory muscle activity.
Summary and conclusion
In recent years researchers have discovered that mechanical ventilation can damage previously injured lungs [68]. Mechanical ventilation can also damage the previously normal respiratory muscles. CMV imposes a unique form of skeletal muscle disuse: the diaphragm is simultaneously unloaded, electrically quiescent, and phasically shortened by cyclical lung inflation, or tonically shortened when PEEP is used. Recent microarray analysis identified 354 differentially expressed gene products in the diaphragms of animals subjected to CMV compared to control animals [69]. Intense research is required to unravel the mechanisms of VIDD and discover ways to translate this knowledge into clinical benefit.
The respiratory muscles are not an inert mechanical pump that can be “light-heartedly” substituted by the ventilator. The respiratory muscles should remain active because they are plastic, and vulnerable.
References
Vassilakopoulos T, Petrof BJ (2004) Ventilator-induced diaphragmatic dysfunction. Am J Respir Crit Care Med 169:336–341
Arroliga A, Frutos-Vivar F, Hall J, Esteban A, Apezteguia C, Soto L, Anzueto A (2005) Use of sedatives and neuromuscular blockers in a cohort of patients receiving mechanical ventilation. Chest 128:496–506
Anzueto A, Peters JI, Tobin MJ, de los SR, Seidenfeld JJ, Moore G, Cox WJ, Coalson JJ (1997) Effects of prolonged controlled mechanical ventilation on diaphragmatic function in healthy adult baboons. Crit Care Med 25:1187–1190
Radell PJ, Remahl S, Nichols DG, Eriksson LI (2002) Effects of prolonged mechanical ventilation and inactivity on piglet diaphragm function. Intensive Care Med 28:358–364
Sassoon CS, Caiozzo VJ, Manka A, Sieck GC (2002) Altered diaphragm contractile properties with controlled mechanical ventilation. J Appl Physiol 92:2585–2595
Jaber S, Sebbane M, Koechlin C, Hayot M, Capdevila X, Eledjam JJ, Prefaut C, Ramonatxo M, Matecki S (2005) Effects of short vs. prolonged mechanical ventilation on antioxidant systems in piglet diaphragm. Intensive Care Med 31:1427–1433
Le Bourdelles G, Viires N, Boczkowski J, Seta N, Pavlovic D, Aubier M (1994) Effects of mechanical ventilation on diaphragmatic contractile properties in rats. Am J Respir Crit Care Med 149:1539–1544
Powers SK, Shanely RA, Coombes JS, Koesterer TJ, McKenzie M, Van Gammeren D, Cicale M, Dodd SL (2002) Mechanical ventilation results in progressive contractile dysfunction in the diaphragm. J Appl Physiol 92:1851–1858
Yang L, Luo J, Bourdon J, Lin MC, Gottfried SB, Petrof BJ (2002) Controlled mechanical ventilation leads to remodeling of the rat diaphragm. Am J Respir Crit Care Med 166:1135–1140
Capdevila X, Lopez S, Bernard N, Rabischong E, Ramonatxo M, Martinazzo G, Prefaut C (2003) Effects of controlled mechanical ventilation on respiratory muscle contractile properties in rabbits. Intensive Care Med 29:103–110
Zhu E, Sassoon CS, Nelson R, Pham HT, Zhu L, Baker MJ, Caiozzo VJ (2005) Early effects of mechanical ventilation on isotonic contractile properties and MAF-box gene expression in the diaphragm. J Appl Physiol 99:747–756
Shanely RA, Zergeroglu MA, Lennon SL, Sugiura T, Yimlamai T, Enns D, Belcastro A, Powers SK (2002) Mechanical ventilation-induced diaphragmatic atrophy is associated with oxidative injury and increased proteolytic activity. Am J Respir Crit Care Med 166:1369–1374
Hussain SN, Vassilakopoulos T (2002) Ventilator-induced cachexia. Am J Respir Crit Care Med 166:1307–1308
McClung JM, Kavazis AN, Deruisseau KC, Falk DJ, Deering MA, Lee Y, Sugiura T, Powers SK (2007) Caspase-3 regulation of diaphragm myonuclear domain during mechanical ventilation-induced atrophy. Am J Respir Crit Care Med 175:150–159
Jarvinen MJ, Einola SA, Virtanen EO (1992) Effect of the position of immobilization upon the tensile properties of the rat gastrocnemius muscle. Arch Phys Med Rehabil 73:253–257
Jokl P, Konstadt S (1983) The effect of limb immobilization on muscle function and protein composition. Clin Orthop Relat Res 222–229
Allen DL, Roy RR, Edgerton VR (1999) Myonuclear domains in muscle adaptation and disease. Muscle Nerve 22:1350–1360
Shanely RA, Van Gammeren D, Deruisseau KC, Zergeroglu AM, McKenzie MJ, Yarasheski KE, Powers SK (2004) Mechanical ventilation depresses protein synthesis in the rat diaphragm. Am J Respir Crit Care Med 170:994–999
Gayan-Ramirez G, De Paepe K, Cadot P, Decramer M (2003) Detrimental effects of short-term mechanical ventilation on diaphragm function and IGF-I mRNA in rats. Intensive Care Med 29:825–833
Powers SK, Kavazis AN, Deruisseau KC (2005) Mechanisms of disuse muscle atrophy: role of oxidative stress. Am J Physiol Regul Integr Comp Physiol 288:R337–R344
Maes K, Testelmans D, Powers S, Decramer M, Gayan-Ramirez G (2007) Leupeptin inhibits ventilator-induced diaphragm dysfunction in rats. Am J Respir Crit Care Med 175:1134–1138
Racz GZ, Gayan-Ramirez G, Testelmans D, Cadot P, De Paepe K, Zador E, Wuytack F, Decramer M (2003) Early changes in rat diaphragm biology with mechanical ventilation. Am J Respir Crit Care Med 168:297–304
Du J, Wang X, Miereles C, Bailey JL, Debigare R, Zheng B, Price SR, Mitch WE (2004) Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J Clin Invest 113:115–123
Deruisseau KC, Kavazis AN, Deering MA, Falk DJ, Van Gammeren D, Yimlamai T, Ordway GA, Powers SK (2005) Mechanical ventilation induces alterations of the ubiquitin–proteasome pathway in the diaphragm. J Appl Physiol 98:1314–1321
Mitch WE, Goldberg AL (1996) Mechanisms of muscle wasting. The role of the ubiquitin–proteasome pathway. N Engl J Med 335:1897–1905
Sassoon CS, Zhu E, Caiozzo VJ (2004) Assist–control mechanical ventilation attenuates ventilator-induced diaphragmatic dysfunction. Am J Respir Crit Care Med 170:626–632
Testelmans D, Maes K, Wouters P, Gosselin N, Deruisseau K, Powers S, Sciot R, Decramer M, Gayan-Ramirez G (2006) Rocuronium exacerbates mechanical ventilation-induced diaphragm dysfunction in rats. Crit Care Med 34:3018–3023
Zergeroglu MA, McKenzie MJ, Shanely RA, Van Gammeren D, Deruisseau KC, Powers SK (2003) Mechanical ventilation-induced oxidative stress in the diaphragm. J Appl Physiol 95:1116–1124
Shringarpure R, Grune T, Davies KJ (2001) Protein oxidation and 20S proteasome-dependent proteolysis in mammalian cells. Cell Mol Life Sci 58:1442–1450
Davies KJ (2001) Degradation of oxidized proteins by the 20S proteasome. Biochimie 83:301–310
Falk DJ, Deruisseau KC, Van Gammeren DL, Deering MA, Kavazis AN, Powers SK (2006) Mechanical ventilation promotes redox status alterations in the diaphragm. J Appl Physiol 101:1017–1024
Reid MB (2001) Invited Review: Redox modulation of skeletal muscle contraction: what we know and what we don't. J Appl Physiol 90:724–731
Hussain SN, Matar G, Barreiro E, Florian M, Divangahi M, Vassilakopoulos T (2006) Modifications of proteins by 4-hydroxy-2-nonenal in the ventilatory muscles of rats. Am J Physiol Lung Cell Mol Physiol 290:L996–1003
Siems W, Capuozzo E, Lucano A, Salerno C, Crifo C (2003) High sensitivity of plasma membrane ion transport ATPases from human neutrophils towards 4-hydroxy-2,3-trans-nonenal. Life Sci 73:2583–2590
Bernard N, Matecki S, Py G, Lopez S, Mercier J, Capdevila X (2003) Effects of prolonged mechanical ventilation on respiratory muscle ultrastructure and mitochondrial respiration in rabbits. Intensive Care Med 29:111–118
Bottinelli R (2001) Functional heterogeneity of mammalian single muscle fibres: do myosin isoforms tell the whole story? Pflugers Arch 443:6–17
Geiger PC, Cody MJ, Macken RL, Sieck GC (2000) Maximum specific force depends on myosin heavy chain content in rat diaphragm muscle fibers. J Appl Physiol 89:695–703
Shanely RA, Coombes JS, Zergeroglu AM, Webb AI, Powers SK (2003) Short-duration mechanical ventilation enhances diaphragmatic fatigue resistance but impairs force production. Chest 123:195–201
Talmadge RJ (2000) Myosin heavy chain isoform expression following reduced neuromuscular activity: potential regulatory mechanisms. Muscle Nerve 23:661–679
Staib JL, Swoap SJ, Powers SK (2002) Diaphragm contractile dysfunction in MyoD gene-inactivated mice. Am J Physiol Regul Integr Comp Physiol 283:R583–R590
Testelmans D, Maes K, Wouters P, Powers SK, Decramer M, Gayan-Ramirez G (2007) Infusions of rocuronium and cisatracurium exert different effects on rat diaphragm function. Intensive Care Med 33:872–879
Watson AC, Hughes PD, Louise HM, Hart N, Ware RJ, Wendon J, Green M, Moxham J (2001) Measurement of twitch transdiaphragmatic, esophageal, and endotracheal tube pressure with bilateral anterolateral magnetic phrenic nerve stimulation in patients in the intensive care unit. Crit Care Med 29:1325–1331
Laghi F, Cattapan SE, Jubran A, Parthasarathy S, Warshawsky P, Choi YS, Tobin MJ (2003) Is weaning failure caused by low-frequency fatigue of the diaphragm? Am J Respir Crit Care Med 167:120–127
Ayas NT, McCool FD, Gore R, Lieberman SL, Brown R (1999) Prevention of human diaphragm atrophy with short periods of electrical stimulation. Am J Respir Crit Care Med 159:2018–2020
Knisely AS, Leal SM, Singer DB (1988) Abnormalities of diaphragmatic muscle in neonates with ventilated lungs. J Pediatr 113:1074–1077
Levine S, Nguyen T, BSE, Friscia M, Kaiser LR, Shrager JB (2006) Ventilator-induced atrophy in human diaphragm myofibers. Proc Am Thorac Soc [Abstract] 3:A27
Nguyen T, Friscia M, Kaiser LR, Shrager JB, Levine S (2006) Ventilator–induced proteolysis in human diaphragm myofibers. Proc Am Thorac Soc [Abstract] 3:A259
Deem S, Lee CM, Curtis JR (2003) Acquired neuromuscular disorders in the intensive care unit. Am J Respir Crit Care Med 168:735–739
Criswell DS, Shanely RA, Betters JJ, McKenzie MJ, Sellman JE, Van Gammeren DL, Powers SK (2003) Cumulative effects of aging and mechanical ventilation on in vitro diaphragm function. Chest 124:2302–2308
Zakynthinos SG, Vassilakopoulos T, Daniil Z, Zakynthinos E, Koutsoukos E, Katsouyianni K, Roussos C (1997) Pressure support ventilation in adult respiratory distress syndrome: short-term effects of a servocontrolled mode. J Crit Care 12:161–172
Putensen C, Zech S, Wrigge H, Zinserling J, Stuber F, Von Spiegel T, Mutz N (2001) Long-term effects of spontaneous breathing during ventilatory support in patients with acute lung injury. Am J Respir Crit Care Med 164:43–49
Ebihara S, Hussain SN, Danialou G, Cho WK, Gottfried SB, Petrof BJ (2002) Mechanical ventilation protects against diaphragm injury in sepsis: interaction of oxidative and mechanical stresses. Am J Respir Crit Care Med 165:221–228
Nava S, Gregoretti C, Fanfulla F, Squadrone E, Grassi M, Carlucci A, Beltrame F, Navalesi P (2005) Noninvasive ventilation to prevent respiratory failure after extubation in high-risk patients. Crit Care Med 33:2465–2470
Ferrer M, Esquinas A, Arancibia F, Bauer TT, Gonzalez G, Carrillo A, Rodriguez-Roisin R, Torres A (2003) Noninvasive ventilation during persistent weaning failure: a randomized controlled trial. Am J Respir Crit Care Med 168:70–76
Ferrer M, Valencia M, Nicolas JM, Bernadich O, Badia JR, Torres A (2006) Early noninvasive ventilation averts extubation failure in patients at risk: a randomized trial. Am J Respir Crit Care Med 173:164–170
Vassilakopoulos T, Zakynthinos S, Roussos C (2005) Bench-to-bedside review: Weaning failure – should we rest the respiratory muscles with controlled mechanical ventilation? Crit Care 10:204
Vassilakopoulos T, Zakynthinos S, Roussos C (1996) Respiratory muscles and weaning failure. Eur Respir J 9:2383–2400
Vassilakopoulos T, Roussos C, Zakynthinos S (1999) Weaning from mechanical ventilation. J Crit Care 14:39–62
Vassilakopoulos T, Zakynthinos S, Roussos C (1998) The tension-time index and the frequency/tidal volume ratio are the major pathophysiologic determinants of weaning failure and success. Am J Respir Crit Care Med 158:378–385
Gayan-Ramirez G, Testelmans D, Maes K, Racz GZ, Cadot P, Zador E, Wuytack F, Decramer M (2005) Intermittent spontaneous breathing protects the rat diaphragm from mechanical ventilation effects. Crit Care Med 33:2804–2809
Betters JL, Criswell DS, Shanely RA, Van Gammeren D, Falk D, Deruisseau KC, Deering M, Yimlamai T, Powers SK (2004) Trolox attenuates mechanical ventilation-induced diaphragmatic dysfunction and proteolysis. Am J Respir Crit Care Med 170:1179–1184
Grundy JE, Storey KB (1998) Antioxidant defenses and lipid peroxidation damage in estivating toads, Scaphiopus couchii. J Comp Physiol [B] 168:132–142
Hudson NJ, Franklin CE (2002) Maintaining muscle mass during extended disuse: aestivating frogs as a model species. J Exp Biol 205:2297–2303
Nathens AB, Neff MJ, Jurkovich GJ, Klotz P, Farver K, Ruzinski JT, Radella F, Garcia I, Maier RV (2002) Randomized, prospective trial of antioxidant supplementation in critically ill surgical patients. Ann Surg 236:814–822
Heyland D (2005) Antioxidant nutrients:a systematic review of trace elements and vitamins in the critically ill patient. Intensive Care Med 31:327–337
Vijayan K, Thompson JL, Norenberg KM, Fitts RH, Riley DA (2001) Fiber-type susceptibility to eccentric contraction-induced damage of hindlimb-unloaded rat AL muscles. J Appl Physiol 90:770–776
Van Gammeren D, Falk DJ, Deruisseau KC, Sellman JE, Decramer M, Powers SK (2005) Reloading the diaphragm following mechanical ventilation does not promote injury. Chest 127:2204–2210
International consensus conferences in intensive care medicine: Ventilator-associated lung injury in ARDS. This official conference report was cosponsored by the American Thoracic Society, The European Society of Intensive Care Medicine, and The Societe de Reanimation de Langue Francaise, and was approved by the ATS Board of Directors, July 1999. Am J Respir Crit Care Med 160:2118–2124
Deruisseau KC, Shanely RA, Akunuri N, Hamilton MT, Van Gammeren D, Zergeroglu AM, McKenzie M, Powers SK (2005) Diaphragm unloading via controlled mechanical ventilation alters the gene expression profile. Am J Respir Crit Care Med 172:1267–1275
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Rights and permissions
About this article
Cite this article
Vassilakopoulos, T. Ventilator-induced diaphragm dysfunction: the clinical relevance of animal models. Intensive Care Med 34, 7–16 (2008). https://doi.org/10.1007/s00134-007-0866-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00134-007-0866-x