

Abstract
Since the early 1970s there has been an ongoing debate regarding the wisdom of promoting unassisted spontaneous breathing throughout the course of critical illness in patients with severe respiratory failure. The basis of this debate has focused on the clinical relevance of opposite problems. Historically, the term “disuse atrophy” has described a situation wherein sustained inactivity of the respiratory muscles (ie, passive ventilation) results in deconditioning and weakness. More recently it has been referred to as “ventilator-induced diaphragmatic dysfunction.” In contrast, “use atrophy” describes a situation where chronic high-tension inspiratory work causes structural damage to the diaphragm and weakness. Both laboratory and clinical studies demonstrated that relatively brief periods of complete respiratory muscle inactivity, as well as intense muscle loading, result in acute inflammation, loss of muscle mass, and weakness. Yet in critical illness other factors also affect respiratory muscle function, including prolonged use of neuromuscular blocking agents, administration of corticosteroids, and sepsis. This makes the attribution of acquired respiratory muscle weakness and ventilator-dependence to either ventilator-induced diaphragmatic dysfunction or loaded breathing extremely difficult. Regardless, the clinical implications of this research strongly suggest that passive mechanical ventilation should be avoided whenever possible. However, promotion of unassisted spontaneous breathing in the acute phase of critical illness also may carry a substantial risk of respiratory muscle injury and weakness. Use of mechanical ventilation modes in a manner that induces spontaneous breathing effort, while simultaneously reducing the work load on the respiratory muscles, is probably sufficient to minimize both problems.
- acute lung injury
- acute respiratory distress syndrome
- discoordinated breathing
- disuse atrophy
- respiratory muscle fatigue
- ventilator-induced diaphragmatic dysfunction
- use atrophy
Introduction
The salutary effects of maintaining spontaneous breathing during critical illness have been debated since the early 1970s, with the advent of intermittent mandatory ventilation (IMV).1 Proponents of IMV claimed that this mode could achieve improved patient-ventilator synchrony, require less need for sedation, and provide faster weaning.2 The conceptual foundation of IMV, and thus a large measure of its physiologic legitimacy, is that continuous spontaneous breathing by patients with acute respiratory failure is beneficial—a claim, however, that was never substantiated.3,4 The invention of airway pressure release ventilation (APRV), in the late 1980s,5 was accompanied by similar claims of improved respiratory muscle function and reduced time on mechanical ventilation,6,7 thus extending the debate surrounding the role of spontaneous breathing during critical illness into contemporary practice. This is hardly surprising, as the inventors of APRV include some of the same investigators who invented and popularized IMV.
In this paper I consider the merits and risks of promoting spontaneous breathing in acute lung injury (ALI) and acute respiratory distress syndrome (ARDS), wherein the focus is the impact on respiratory muscle function. To alert the reader, I will use the general term “respiratory muscles” for simplicity of writing, yet in most instances the emphasis is on inspiratory muscle function, and in particular diaphragmatic function. In addition, over the past 40 years the description of respiratory muscle activity during mechanical ventilation has resulted in an array of terms that often confuses these discussions.8 In particular, the various interpretations associated with the term “controlled mechanical ventilation” is problematic. It possesses legitimacy primarily because of its historical currency. Thus, it is difficult to decipher 4 decades of literature on the evolution of mechanical ventilation without referring to the term.
In the context of this paper, however, the term “passive mechanical ventilation” is used instead to denote mechanical ventilation that occurs in the absence of spontaneous breathing efforts, regardless of whether or not this condition is pharmacologically induced. Thus, passive mechanical ventilation will subsume the use of terms such as volume, pressure, and dual-mode control, as well as IMV, when respiratory muscle activity is absent. However, the term “assisted mechanical ventilation” is retained for convenience and refers to conditions wherein spontaneous breathing efforts are synchronized with significant degrees of mechanical support. This would include high levels of pressure support ventilation and even IMV at a frequency sufficient to minimize unsupported, spontaneously generated breaths
Respiratory Muscle Function During Critical Illness
The controversy over the role of spontaneous breathing during critical illness centers on the clinical relevance of opposite problems. Simply stated, sustained, inactivity of the respiratory muscles results in loss of muscle mass9 and strength,10 whereas the sustained imposition of high-tension work causes structural damage11–13 and may exacerbate inflammation of the diaphragm.14 This conundrum has been described as “disuse” versus “use atrophy”13; both conditions may contribute to prolonged ventilator-dependence in patients with severe respiratory failure and indirectly may impact clinical outcomes.
Discoordinated Breathing
Discoordinated breathing was a prominent and early justification for promoting spontaneous breathing throughout the course of critical illness. The origins of this controversy began in the early 1970s, with the observation that approximately 5% of patients recovering from acute respiratory failure showed signs of respiratory muscle “discoordination,” of varying severity, that could not be attributed to underlying (ie, primary) neuromuscular disease.15 Discoordinated breathing first was described briefly in a larger paper on mechanical ventilation and was defined as the onset of expiratory activity while the chest cage was still expanding.15 It was considered to be idiopathic, was observed to resolve slowly, and in consequence delayed weaning.
However, a subsequent, detailed observational study attributed discoordinated breathing to the effects of hyperinflation on diaphragmatic motion in patients with severe COPD.16 It should be noted that this breathing pattern was described prior to the discovery of intrinsic PEEP, with its associated threshold loading, uncaptured inspiratory efforts, and recruitment of abdominal expiratory muscles that also might contribute to the clinical impression of discoordinated breathing.17,18
Yet proponents of IMV emphasized discoordinated breathing and prolonged weaning following passive mechanical ventilation as a justification for promoting spontaneous breathing throughout the course of mechanical ventilation. They claimed that “almost all patients requiring ventilatory support for more than 24 hours develop discoordination of abdominal and accessory muscles of respiration” that “may prolong dependence on the ventilator” and “does not occur in patients who breathe spontaneous throughout their treatment with IMV.”2 Although discoordinated breathing was not attributed directly to muscle deconditioning from use of passive mechanical ventilation,2 the linkage of these 2 phenomena was strongly implied. A small observational study19 of 14 patients (6 with COPD and 8 with acute respiratory failure and no evidence of preexisting lung disease) seemed to support that claim. Unfortunately, that study never was published beyond abstract form, so that detailed information on the relationship between discoordinated breathing and respiratory muscle fatigue, or deconditioning in those patients with acute respiratory failure was not forthcoming.
Respiratory Muscle Fatigue
Like other skeletal muscles, the diaphragm is subject to fatigue,20 defined as loss of a muscle's capacity for developing force and/or velocity, resulting from activity under loaded conditions, and which is reversible with rest.21 For the respiratory system, that muscular force translates into the pleural pressure necessary to sustain minute ventilation at a level achieving eucapnia. Fatigue is distinguished from muscle weakness, as the latter denotes the inability to develop a targeted force in a rested muscle.21
Unlike other skeletal muscles, in which rest can reverse fatigue, the diaphragm must remain perpetually active to sustain life. As the primary muscle of ventilation it also must be capable of extremely high work output to accomplish sneezing, coughing, and bodily exertion. The solution to this unique problem is reflected in the proportional composition of muscle fibers. Approximately 55% of the diaphragm consists of Type 1 (“slow twitch”) muscle fibers that are highly resistant to fatigue, but generate relatively low levels of force.13,21,22 These fibers represent the portion of the diaphragm that is perpetually active during normal breathing. The remaining 45% of the diaphragm consists of 2 fibers that are sequentially recruited in proportion to increasing levels of minute-ventilation demand and/or work of breathing. This muscle tissue consists of “fast twitch” Type 2a (intermediate fatigue-resistant/intermediate force-generating capacity) and Type 2b (low fatigue-resistant/highest force-generating) fibers.21,22 The accessory muscles of respiration also posses a similar composition of fiber types.21,22
This structural composition of the respiratory muscles suggests a time-limited work level under loaded conditions whereby minute ventilation can be sustained. Under these conditions, hypercapnia represents either fatigue of the inspiratory muscles or an adaptive strategy to preserve some degree of muscular function so as to prevent overt failure.21,23
Fatigue is defined further as either “low frequency” or “high frequency,” according to the muscle fibers involved and the duration of degraded muscle performance.24 In addition, the descriptor “incipient fatigue” has been used to describe situations in which voluntary respiratory muscle contractions are limited by central inhibition in the face of excessive work demands, but external electrical diaphragmatic stimulation is capable of eliciting stronger contractions.25 In contrast, “overt fatigue” refers to the inability of external electrical stimulation to evoke stronger respiratory muscle contractions.25 During normal tidal breathing, respiratory muscles are normally stimulated at a frequency range of 10–20 Hz, whereas stimulation at higher frequencies (eg, 60–100 Hz) result in more forceful contractions.26 High-frequency fatigue, as it involves fast-twitch fibers and forceful contractions, is believed to be caused by the accumulation of inorganic phosphate and the failure of electrical conduction to the contractile fibers in the muscle, as well as intramuscular acidosis.24 Recovery from high-frequency fatigue generally occurs within 15 min.24 In contrast, low-frequency fatigue is believed to be caused by muscle-fiber injury, recovery from which may take several days.24
Diaphragmatic Function During Loaded Breathing
Excessive loading of the inspiratory muscles induces acute fatigue,20,27–33 from which complete recovery requires 24–48 hours, even when that load is experienced for relatively brief periods.13,34,35 At functional residual capacity the inspiratory muscles can sustain increased work loads indefinitely, as long as the inspiratory change in transdiaphragmatic pressure (ΔPdi) is less than 40% of maximum.28 Beyond this, fatigue eventually ensues at a time point inversely related to magnitude of the load.28 When subjects are allowed to recruit all of their inspiratory muscles, fatigue-onset occurs when the imposed work load increases to between 50–70% of the maximal inspiratory pressure.27
Furthermore, the time to fatigue-onset also depends upon the percentage of the respiratory cycle committed to inspiration.30 For any ΔPdi, time to fatigue-onset decreases as inspiratory time increases. This is represented by the tension-time index of the diaphragm, which is the product of the ratio of ΔPdi to maximum Pdi and percent-inspiratory time. That is:
in which TTIdi signifies respiratory muscle oxygen consumption,30 as well as reflecting limitations in muscle perfusion that affect oxygen supply, removal of metabolites, and maintenance of local electrolyte balance.31 A value exceeding 0.15 is considered the critical cut-off value for the development of fatigue.30
Of particular interest is the presence of “discoordinated breathing” during diaphragmatic fatigue. In normal subjects at fatigue-onset, the breathing pattern has been observed to become “irregular and disorganized,” whereby inspiration is accomplished in steps (“cogwheel pattern”).20 When all inspiratory muscles are allowed to function during fatiguing loads in normal subjects, over time the partitioning of work alternates between the diaphragm and the intercostals/accessory muscles (“respiratory alterans”).27 This gives the distinct impression of “discoordination”36 that becomes more salient with pronounced recruitment of the abdominal muscles during expiration.27 Respiratory alterans has been observed clinically in patients with inspiratory muscle fatigue, including those with ALI/ARDS,29 so that the perception of “discoordinated breathing” may signify impending fatigue from excessive muscle loading rather than deconditioning per se.
Reversal of muscle fatigue requires rest. However, it is uncertain in clinical practice whether this necessitates completely “shutting-down” the inspiratory muscle with passive mechanical ventilation, or just reducing the power of breathing to normal or subnormal levels for a certain period of time.13,21 For example, following induction of diaphragmatic fatigue in normal subjects, recovery was 75% complete within 3 hours and 100% complete by 25 hours with subjects spontaneously breathing at normal work loads.34 By extension, the failure to adequately reduce inspiratory muscle work load suggests that a state of chronic respiratory muscle fatigue may occur.13,23 Proponents of IMV have advocated use of a mandatory rate that “will just prevent acidemia.”2 This raises a concern that prolonged ventilator-dependence following acute respiratory failure may, in part, be a consequence of insufficient mechanical support. In some circumstances chronic fatigue may persist for several days or even weeks.13 How much time and what degree of mechanical ventilatory support is required for inspiratory muscle recovery following critical illness is unknown and remains speculative.13,21 It should be noted that muscle fatigue is distinct from muscle weakness; however, weak muscles are more susceptible to fatigue21 because the maximum inspiratory force reserve is diminished.
Finally, there is considerable evidence from patients with chronic lung disease and inspiratory muscle weakness that several hours of mechanical ventilation use each day (usually nocturnal) markedly improve inspiratory muscle function.13,23,37–41 This supports the suggestion that allowing partial or complete unloading of the inspiratory muscles for a period of 4–10 hours a day may play a direct role.13 However, the mechanism for improvement in these patients is complex, and improved respiratory muscle function has not been a universal finding.42 This has led to an alternative, and perhaps complementary, theory that noninvasive ventilation in these patients improves sleep quality, thus preventing chronic nocturnal hypoventilation that exacerbates blunted respiratory drive and in consequence improves nervous-system responsiveness to carbon dioxide.43
Loaded Breathing and Diaphragmatic Injury
As mentioned earlier, sustained high-tension work causes structural damage to and inflammation of the diaphragm.11–14 Exhaustive contractions in “untrained” skeletal muscle causes structural damage as well as degenerative changes in muscle tissue that persists for a week following the event.44 This appears to be more prominent in highly oxidative, fatigue-resistant (Type 1) fibers.44 The associated inflammatory response has been shown to persist for several days in human skeletal muscle.45 Delayed inflammation or secondary injury to both the diaphragm and parasternal intercostal muscles has been demonstrated 3 days following brief periods of fatigue-inducing inspiratory resistive loading.26,46 Moreover, delayed injury appears to increase the susceptibility of the respiratory muscles to repeated injury.26
Structural damage to the respiratory muscles from loaded breathing also causes oxidative stress and stimulates pro-inflammatory cytokine production.47 Normal subjects breathing at 75% of their maximal inspiratory pressure for less than one hour have significant elevations in plasma levels of tumor necrosis factor alpha, interleukin-1B and interleukin-6,48 as well lymphocyte activation.47 Tumor necrosis factor alpha, also has been shown to reduce diaphragmatic force generation.26
In dogs, high-load inspiratory resistive breathing below the fatigue level (ie, tension-time index of 0.12) for 2 hours a day produced cell membrane disruption and sarcomere damage primarily to Type-1 fibers.11 Diaphragmatic tissue biopsies from elective thoracic surgery patients exposed to brief periods of fatigue-induced threshold-loaded breathing were found to have sarcomere damage.12 Interestingly, the damage was more pronounced in patients with chronic lung disease. It was speculated that the greater susceptibility to damage observed in these patients may have resulted from in-coordination or co-contraction of agonist and antagonistic muscle groups, or from eccentric loads (ie, differential contraction intensity between diaphragmatic sub-segments).49 Three days following exposure to a brief period (1.5 h) of fatiguing inspiratory resistive loads (ie, tension-time index of 0.22), rabbits exhibited a marked increase in shredded and necrotic diaphragmatic fibers as well as inflammatory cell infiltration.26,46 Ischemia causes increased reactive oxygen species (ROS) production that, in turn, stimulates cellular proteolytic enzyme systems, leading to muscle degradation. In those rabbits the proteolytic calpain system was activated.
When assessing the risk of muscle fiber damage from loaded breathing in the context of ALI/ARDS, it is interesting to note that the application of passive mechanical ventilation in animals with experimental sepsis actually eliminated cell damage and improved diaphragmatic performance.50 This is most likely explained by the fact that sepsis induces sarcolemma damage and respiratory muscle weakness, so that loaded breathing probably aggravates the damage further.14
Ventilator-Induced Diaphragmatic Dysfunction
Until recently, very little research had been done on respiratory muscle weakness as a result of disuse muscle atrophy. Disuse atrophy of skeletal muscles was demonstrated in physiology experiments over 70 years ago.51 Since then, numerous clinical studies have affirmed that prolonged skeletal muscle inactivity results in atrophy, weakness, and neuromuscular impairment.52–54 In brief, skeletal muscle disuse decreases muscle volume and weight with a corresponding loss of total muscle contractile proteins, as well as mitochondria and sarcoplasmic reticulum that impairs energy production and calcium activity, respectively.54 Disuse atrophy is caused by decreased protein synthesis and/or increased proteolysis within muscle tissue.55 More active skeletal muscles have a higher propensity to develop atrophy from prolonged inactivity.56 As the diaphragm maintains a constant activity level between 30–40%, it appears to be particularly susceptible to disuse atrophy.57
Ventilator-induced diaphragmatic dysfunction (VIDD) is defined as the loss of diaphragmatic force-generating capacity specifically related to the use of passive mechanical ventilation.55 It is characterized by structural damage to muscle fibers from oxidative stress, as well as muscle atrophy. Evidence from animal models of VIDD suggests that several pathways are involved.57 Prolonged diaphragmatic quiescence results in both decreased protein synthesis and increased muscle tissue proteolysis.55 Whereas decreased production of insulin-like growth factor-1 (IGF-1) reduces the expression of protein kinases responsible for protein synthesis,57 the genesis of structural damage to the diaphragm is more complex (Fig. 1).

Cellular mechanisms implication in ventilator-induced diaphragmatic dysfunction. Bid = BH3-interacting death domain protein. NADPH reduced nicotinamide adenine dinucleotide phosphate.
Diaphragmatic muscle fiber damage results from increased levels of ROS.55 However, this is not associated with either inflammatory cell infiltration or increased production of pro-inflammatory cytokines. Damage to the mitochondria appears to be a significant source of ROS.57 The other major source of ROS comes from activation of cellular proteolytic systems that also are activated by increased levels of ROS. Of these, the mutually reinforcing caspase and calpain systems are believed to drive ROS production, resulting in protein degradation,55,57 the primary targets being the contractile elements of the diaphragm (ie, actin and myosin). Furthermore, activation of the caspase and calpain systems may activate the protein Bid (BH3-interacting death domain protein), causing further mitochondrial damage, and thus potentiating ROS production.57
As an area of scientific inquiry, VIDD is relatively new, with the first controlled study published in 1994, wherein rats exposed to passive ventilation for 48 hours had significantly reduced diaphragmatic weight and force generation.58 Since that time, numerous animal studies confirmed that passive mechanical ventilation results in disuse atrophy.10,56,59–62 Rats passively ventilation for 18 hours exhibited diaphragmatic oxidative injury,60 whereas passive mechanical ventilation of 12–24 hours resulted in 35–48% reductions in diaphragmatic twitch force generation.59 Similar findings of maximal Pdi reductions (37–51%) have been reported in rabbits ventilated from 24–72 hours, respectively.62 The magnitude of VIDD differs according to species. For example, baboons passively ventilated for 11 days produced less severe impairment of diaphragmatic function (25% reduction in maximum Pdi),10 whereas piglets passively ventilated for 5 days had only a 20% decrease in maximal Pdi.56 Apparently, larger species require longer periods of inactivity to induce diaphragmatic dysfunction.63
In animal models, VIDD is attenuated substantially either by allowing brief periods of unassisted spontaneous breathing, or use of assisted mechanical ventilation. For example, rats allowed to breathe spontaneously for as little as 5 minutes every 5 hours of passive mechanical ventilation did not exhibit histologic evidence of disuse atrophy and had less deterioration in diaphragmatic force reduction than rats exposed to completely passive ventilation.64 Likewise, rabbits allowed to trigger assisted mechanical breaths for 3 days exhibited substantially less diaphragmatic force reduction, compared to those exposed to passive mechanical ventilation (14% vs 48%, respectively).65
Until recently, the only evidence for VIDD in critically ill humans was a case series from pediatric literature,66 wherein autopsy examinations from 13 neonates who received mechanical ventilation for at least 12 days prior to death reported extensive diaphragmatic atrophy. This was contrasted to the absence of diaphragmatic atrophy in autopsy examinations from 26 infants who were ventilated for up to one week. Recently, however, marked diaphragmatic atrophy was reported in autopsy examination of 14 brain-dead patients following passive mechanical ventilation of only 18–69 hours.9 However, those subjects may not be representative of critically ill patients in general, because brain death is associated with cellular changes that may induce oxidative stress resulting in loss of muscle mass.67 Moreover, the complete loss of neural impulses to the diaphragm also may influence the development of atrophy.68
Respiratory Muscle Dysfunction and Critical Illness Polyneuropathy
Clinically, there are many causes of diaphragmatic weakness and injury during critical illness, so that VIDD is a diagnosis of exclusion.63,69 Other causes include prolonged administration of aminosteroidal neuromuscular blocking agents (pancuronium and vecuronium),70 as well as benzolisoquinoline agents (atracurium).71 This effect can be potentiated by co-administration of high-dose corticosteroids.72 Determining the cause of diaphragmatic weakness is complicated further because it is a common finding in patients suffering from critical illness polyneuropathy,73 which is a pervasive phenomenon in patients with sepsis and multi-organ system dysfunction.74 Approximately 40–50% of patients who develop ARDS have sepsis as the primary risk factor,75–78 which makes the contribution of VIDD to morbidity in patients with ALI/ARDS difficult to determine. Although neuropathies and myopathies associated with critical illness are distinct, clinically the differential diagnosis is difficult.74 Nonetheless, critical-illness myopathy is thought to be as prevalent as critical-illness neuropathy in patients requiring prolonged (> 1 week) stay in the intensive care unit (ie, approximately 58%) and often they may coexist.74
Ventilator-Dependence and Assisted Modes of Mechanical Ventilation
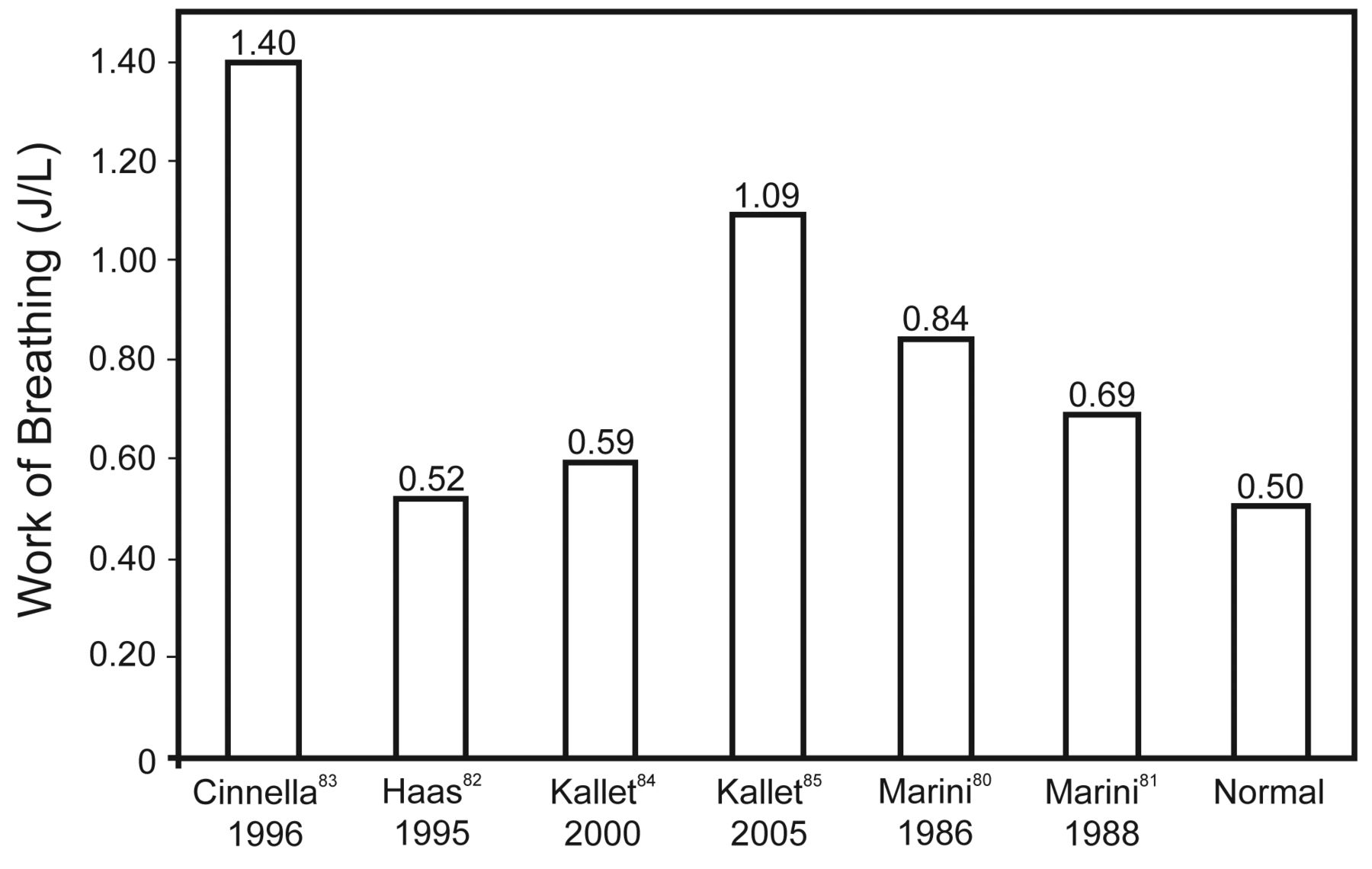
Recent research interest into VIDD has raised an understandable concern regarding appropriate mode selection for mechanical ventilation. Yet VIDD is difficult to diagnose, and its practical impact on the duration of mechanical ventilation is virtually impossible to gauge. A potential source of misunderstanding emanates from conflating the potential dangers of passive mechanical ventilation to modes such as volume control and pressure control ventilation when a patient's spontaneous efforts trigger the majority of breaths. This important distinction is something proponents of IMV and APRV often omit from their arguments. It is unlikely that management of patients with ALI/ARDS using assisted mechanical ventilation adversely affects weaning to any appreciable degree, because patients continue to perform normal to super-normal work of breathing once they initiate assisted breaths from the ventilator (Fig. 2).79–84 Therefore, the risk of VIDD during assisted mechanical ventilation may not be substantial, provided that clinicians are careful to ensure the resumption of assisted mechanical ventilation at the earliest time possible following stabilization of gas-exchange function at reasonable levels of mechanical support.
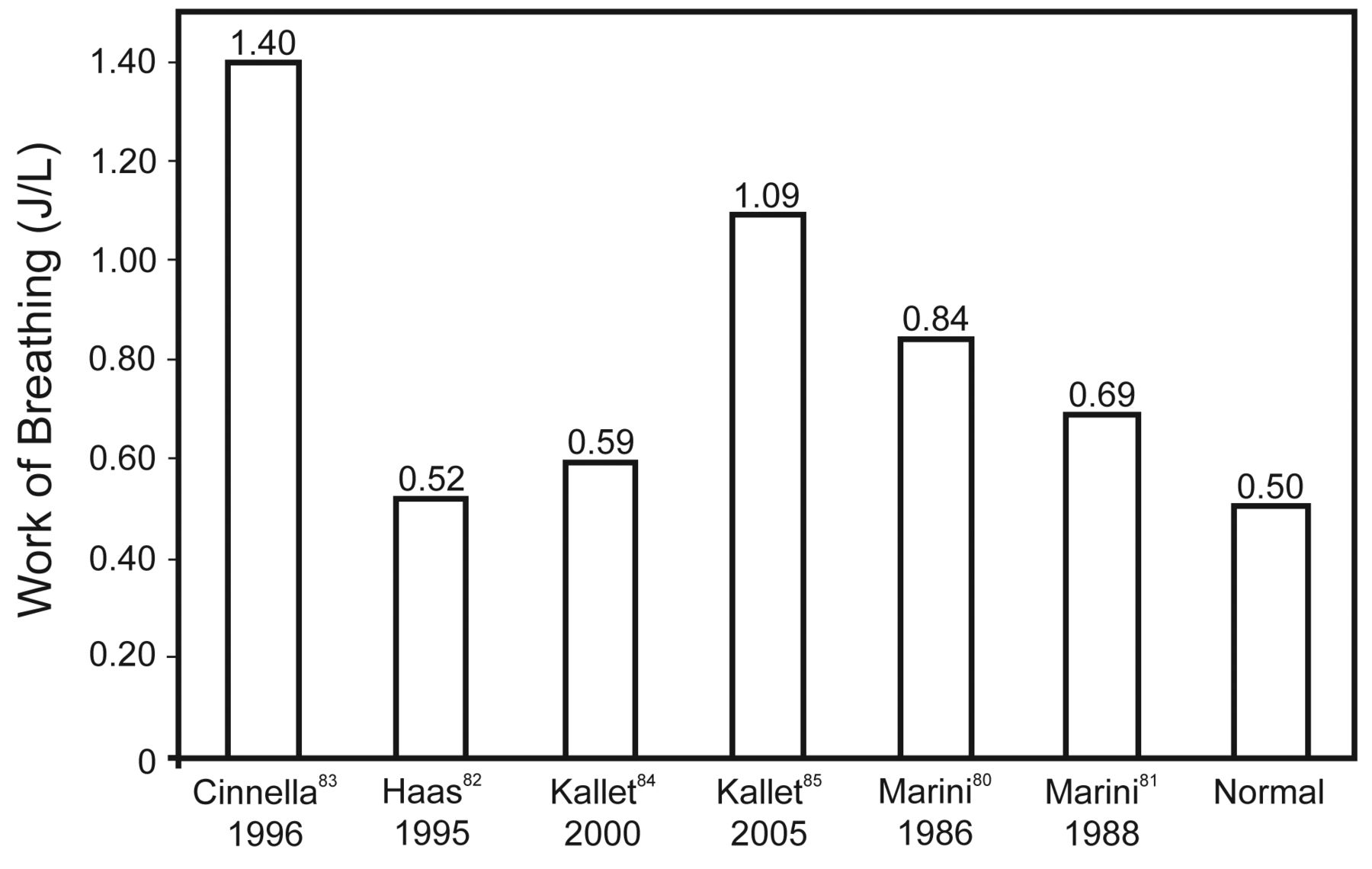
Mean work of breathing values from studies of assisted mechanical ventilation in critically ill patients.
Relative Muscle Weakness and Ventilator-Dependence
The clinical implications of respiratory muscle weakness during critical illness are difficult to assess. In regards to respiratory muscle force production, under laboratory conditions normal humans can generate a maximal Pdi in the range of 140–285 cm H2O,20,30,32,34,35 mean maximal esophageal pressure of 125 cm H2O,32 and mean maximal mouth pressure of 125–145 cm H2O.27,33 In contrast, patients recovering from various causes of respiratory failure, including ALI/ARDS, typically can generate a maximum inspiratory pressure between 30–50 cm H2O.85–90 Despite the acknowledged imprecision associated with the measurement, due to both variations in technique and degree of patient cooperation,85,86 the stark departure from normal suggests that some degree of impaired muscle function is likely. Yet many of these patients are successfully weaned with a maximal inspiratory pressure of only 36–48 cm H2O,87–89 which is approximately 27–36% of normal.
These findings seem to detract from the clinical importance of iatrogenic muscle weakness caused either by chronic excessive muscle loading or VIDD. That relative muscle strength or weakness does not appear to be the primary driver for weaning success may be explained by several factors interacting in complex ways. For example, a low minute ventilation demand coupled with moderately abnormal chest compliance and airways resistance may allow a patient with weak respiratory muscles to sustain spontaneous ventilation, whereas a patient with very abnormal chest mechanics and a high minute ventilation demand may remain ventilator-dependent despite possessing normal respiratory muscle strength. Despite the apparent negative impact that either loaded breathing or VIDD may have on respiratory muscle function, the majority of critically ill patients managed with assisted mechanical ventilation appear to wean without much difficulty,91 despite the fact that many have evidence of relative muscle weakness. Nonetheless, the issue of either chronic excessively loaded breathing or VIDD is likely to be of more clinical relevance in the minority of patients who are most debilitated due to severe chronic disease or a prolonged disease course of particular severity.
Summary
Despite substantial laboratory and clinical evidence linking both severely loaded breathing and prolonged inactivity with respiratory muscle damage and weakness, the practical impact of either problem on the duration of mechanical ventilation, particularly in patients with ALI/ARDS, cannot be discerned. This is because the etiology of respiratory muscle weakness in patients undergoing prolonged mechanical ventilation is probably multi-factorial (Fig. 3). Nonetheless, it would seem prudent to curtail unassisted spontaneous breathing early in the course of critical illness if minute ventilation demand is high and there is significant impairment to chest mechanics. On the other hand, passive mechanical ventilation should be avoided, except for the most severe manifestations of respiratory failure. Whenever high levels of assisted ventilation are required, the mandatory rate should be adjusted to assure patients continue to trigger assisted breaths, whereas sedation should be titrated to the lowest level that promotes reasonable patient-ventilator synchrony without suppressing respiratory drive.

Potential causes of respiratory muscle weakness in mechanically ventilated patients recovering from critical illness.
Footnotes
- Correspondence: Richard H Kallet MSc RRT FAARC, Respiratory Care Services, San Francisco General Hospital, NH:GA-2, 1001 Potrero Avenue, San Francisco CA 94110. E-mail: rkallet{at}sfghsom.ucsf.edu.
-
Mr Kallet has disclosed relationships with Viasys, Covidian, and Puritan Bennett.
-
Mr Kallet presented a version of this paper at the 46th Respiratory Care Journal Conference, “Patient-Ventilator Interaction,” held March 19-21, 2010, in Cancún, Quintana Roo, Mexico.
-
The discussion transcript from Mr Kallet's presentation appears at the end of his second paper to this conference: Kallet RH. Patient-ventilator interaction during acute lung injury, and the role of spontaneous breathing: part 2: airway pressure release ventilation. Respir Care 2011;56(2):190-203; discussion 203-206.
- Copyright © 2011 by Daedalus Enterprises Inc.
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.
- 39.
- 40.
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.
- 77.
- 78.↵
- 79.↵
- 80.
- 81.
- 82.
- 83.
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.
- 89.↵
- 90.↵
- 91.↵





{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Introduction
- Respiratory Muscle Function During Critical Illness
- Discoordinated Breathing
- Respiratory Muscle Fatigue
- Diaphragmatic Function During Loaded Breathing
- Loaded Breathing and Diaphragmatic Injury
- Ventilator-Induced Diaphragmatic Dysfunction
- Respiratory Muscle Dysfunction and Critical Illness Polyneuropathy
- Ventilator-Dependence and Assisted Modes of Mechanical Ventilation
- Relative Muscle Weakness and Ventilator-Dependence
- Summary
- Footnotes
- References
- Figures & Data
- Info & Metrics
- References