

Abstract
Prolonged breathing of very high FIO2 (FIO2 ≥ 0.9) uniformly causes severe hyperoxic acute lung injury (HALI) and, without a reduction of FIO2, is usually fatal. The severity of HALI is directly proportional to PO2 (particularly above 450 mm Hg, or an FIO2 of 0.6) and exposure duration. Hyperoxia produces extraordinary amounts of reactive O2 species that overwhelms natural anti-oxidant defenses and destroys cellular structures through several pathways. Genetic predisposition has been shown to play an important role in HALI among animals, and some genetics-based epidemiologic research suggests that this may be true for humans as well. Clinically, the risk of HALI likely occurs when FIO2 exceeds 0.7, and may become problematic when FIO2 exceeds 0.8 for an extended period of time. Both high-stretch mechanical ventilation and hyperoxia potentiate lung injury and may promote pulmonary infection. During the 1960s, confusion regarding the incidence and relevance of HALI largely reflected such issues as the primitive control of FIO2, the absence of PEEP, and the fact that at the time both ALI and ventilator-induced lung injury were unknown. The advent of PEEP and precise control over FIO2, as well as lung-protective ventilation, and other adjunctive therapies for severe hypoxemia, has greatly reduced the risk of HALI for the vast majority of patients requiring mechanical ventilation in the 21st century. However, a subset of patients with very severe ARDS requiring hyperoxic therapy is at substantial risk for developing HALI, therefore justifying the use of such adjunctive therapies.
- acute lung injury
- acute respiratory distress syndrome
- hyperoxia
- oxygen toxicity
- reactive oxygen species
- ventilator-induced lung injury
Introduction
Though dephlogisticated air might be very useful as a medicine, it might not be so proper for us in the usual healthy state of the body: for, as a candle burns out much faster in dephlogisticated than in common air, so we might, as may be said, live out too fast, and the animals powers be too soon exhausted in this pure kind of air.
— Joseph Priestley1 (1775)
The discovery of O2 in the late 18th century and its linkage to metabolism quickly raised speculation about its potential use for treating cardiopulmonary disease. And almost simultaneously there were concerns that breathing pure O2 might cause irreparable harm and death. In the 1960s, the emergence of the ICU and prolonged mechanical ventilation, along with the increasing use of hyperbaric O2 therapy, made hyperoxia a prominent clinical concern. Even now, nearly 240 years after the discovery of O2, what constitutes the medically safe upper limits and duration of inspired O2 fraction (FIO2) remains uncertain.
In this review we focus on pulmonary O2 toxicity under normobaric conditions. We will provide a selective overview on the history of O2 toxicity research that will focus on questions relevant to acute care practitioners. Then, we will analyze the 50 year debate over the clinical relevance of what is now referred to as “hyperoxic acute lung injury” (HALI): a term that will be used to describe the pulmonary-specific toxic effects of O2.2 Finally, we will describe some of the progress in our understanding of HALI afforded by recent advances in molecular biology. This should help explain both the laboratory and clinical observations of O2 toxicity presented in this paper.
Animal Models of HALI
That O2 was found to be an incendiary invited speculation that breathing it in high concentrations would likely cause lung damage. In 1783 Antoine Lavoisier conducted experiments on guinea pigs that confirmed that breathing an FIO2 of 1.0 led to death from “violent inflammation” and “fiery fever.”1 His postmortem examinations found “the right heart bluish and engorged,” and “the lung was very, very red, stiff and engorged with blood.”3 Lavoisier proposed incorrectly that O2 caused inflammation by increasing pulmonary combustion.4 By 1796 Beddoes and Watt, early advocates of O2 therapy, also had reported marked inflammation in a kitten breathing an FIO2 of 0.8.1,3 By 1866 Jean Baptiste Dumas had published the first study on prolonged breathing at an FIO2 of 1.0. Post-mortem examination of dogs revealed the thorax “filled with acrid serum and coagulated blood; the bronchial tubes were filled with fluid” and “the lungs … had become considerably solidified as occurs in the case of organs which have been for some time inflamed.”3
Prolonged Exposure to Toxic Levels of O2 and Mortality
Since then, virtually all experiments found that most animals die after several days breathing an FIO2 ≥ 0.8. A comprehensive review from the 1970s5 listed over 50 laboratory studies involving 9 animal species (mice, rats, chickens, guinea pigs, rabbits, cats, dogs, monkeys, baboons) and over 1,300 individual animals who breathed an FIO2 of 0.9–1.0 until death. In general, these animals developed progressive respiratory distress and typically died from respiratory failure between 3 to 6 days. Although these studies were done primarily in the 1950s and 1960s,5 the results were consistent with numerous other studies from the 19th and early 20th centuries described in earlier comprehensive reviews.1,3
The time course to death from breathing an FIO2 of 0.9–1.0 extends over a wide range, and is dependent upon the age of the animal, the species, and even the strain. Moreover, individual animals exhibit considerable variability in the time course of developing HALI and subsequent death. Curiously, poikilothermic animals (amphibians and reptiles) seemingly are immune to HALI when their environmental temperature is normal (21°C), having survived up to 52 days breathing an FIO2 of 1.0.1,5–7 However, when the temperature was raised to 38°C, these animals also developed respiratory distress and died from respiratory failure.1 This suggested that HALI is influenced by metabolism.
Relative Toxicity: The Effects of Exposure Time and Concentration
Is there a relatively safe FIO2 that can be used therapeutically for extended periods of time? Regnault and Reiset (1849) were the first to report that several animal species (rabbits and dogs) apparently could breathe an FIO2 of 0.6 indefinitely without signs of toxicity.3 An essential discovery was made in 1878 by physiologist Paul Bert, whose hyperbaric experiments demonstrated that toxicity is caused by the partial pressure of O2.3 This facilitated the earliest conjecture regarding a “safe” upper limit of FIO2. In 1885, EW Moir (chief engineer of the Hudson River tunnel) observed that mules continuously exposed for months at a time to an FIO2 of 0.21 at 30 pounds atmospheric pressure (ie, 450 mm Hg, the equivalent of an FIO2 of 0.6 at 1 atmosphere), showed no signs of toxicity.6 Likewise, in the late 1930s simulated high-altitude experiments in animals breathing an FIO2 of 1.0 at 3,500 m (0.6 atmospheres) also could be endured without ill effect.3
One of the most influential laboratory studies was done in 1899 by James Lorrain Smith,4 who found that mice breathing an FIO2 of 0.4 for over a week exhibited no evidence of toxicity. In contrast, half of the mice exposed to an FIO2 of 0.7–0.8 died of respiratory failure. He concluded that prolonged exposure to an FIO2 of 0.7 likely represents the threshold of substantial toxicity, and at an FIO2 of 0.8 the toxic effects of O2 “varies according to the resistance of the individual animal.”4
Over the next 40 years, several studies essentially confirmed Smith's findings that prolonged exposure to an FIO2 above 0.7 was unambiguously toxic to at least 7 different animal species (mice, rats, birds, guinea pigs, rabbits, cats, and dogs).1,3 Whereas rabbits could breathe at an FIO2 of 0.6 for 3 months without signs of toxicity,1,3,5 prolonged exposure to an FIO2 of 0.7 produced highly variable results, ranging from no distress to death from acute respiratory failure.1 Likewise, sustained exposure to an FIO2 of 0.75–0.9 produced similar pulmonary lesions to that of animals breathing an FIO2 of 0.95–1.0.1 The only pertinent distinction was the delayed onset of toxicity in animals breathing at the lower FIO2. Thus, the general impression from numerous experiments during the first half of the 20th century was that toxicity rose more rapidly as FIO2 was increased above 0.6, and also as exposure time was prolonged.3
Acclimatization to and Recovery From HALI
Other important issues are whether animals can acclimatize to hyperoxia, and whether HALI is reversible. The early evidence reviewed by Bean1 was ambiguous; he concluded that an adaptive response either was absent in many individuals, or that the observed variation in effectiveness was so broad that it likely reflected a process of natural selection.1 Kaplan et al8 reported that most monkeys who survived exposure to an FIO2 of 1.0 for over a week showed distinct signs of adaptation, and by the ninth day respiratory distress began to subside. Monkeys allowed to recover fully prior to sacrifice essentially had normal lungs on postmortem exam. Consistent with previous studies, these investigators also noted the wide variability in response to HALI among individual monkeys, suggesting that genetic predisposition may influence both tolerance and susceptibility to hyperoxia. This is supported by recent evidence from positional cloning studies in mice, in which nuclear transcription factor NRF2 was identified as a gene associated with increased susceptibility to HALI, and that may play a role in human susceptibility to ALI.9
HALI is attenuated by prior, repeated, time-limited exposure to breathing an FIO2 of 1.0,5 whereas injury is enhanced by immunosuppression. Both immunosuppressed mice and mice without prior exposure to hyperoxia had markedly higher rates of pulmonary edema formation.10 Furthermore, immunosuppressed mice uniformly died more quickly, suggesting that an immune response is partly responsible for the ability of some mammals to adapt, at least temporarily, to a high FIO2 environment.
Rats can adapt to prolonged exposure to an FIO2 of 0.85.5,11 This primarily involves the proliferation and hypertrophy of alveolar type II cells possessing larger and more numerous mitochondria, as well as increased antioxidant enzymatic activity. The importance of these adaptive changes may include continued production and secretion of surfactant,11 as hyperoxia has been shown to adversely affect surfactant production.12 Type II cell hyperplasia also may compensate for the direct inhibition of mitochondrial enzymes by hyperoxia.11
Finally, abruptly removing acclimatized animals (eg, dogs, rabbits, monkeys, baboons) from a hyperoxic environment to room air results in rapid onset of cyanosis, respiratory distress, and death.1,8,13,14 This has been attributed to adaptive proliferation or “cellularity” of the alveolar epithelium5,8 that allows survival but creates a pronounced diffusion defect at a substantially lower FIO2.8 Epithelial thickness has been reported to increase by 60% after 72 hours with an associated doubling in “diffusion resistance.”5 Others have reported a 58% decrement in carbon monoxide diffusing capacity in baboons after a week breathing an FIO2 of 1.0.15 Full recovery required gradual weaning of FIO2 over several weeks.8,14 A similar recover trajectory has been reported in humans surviving suspected HALI, whereby recovery was not clinically appreciable until the FIO2 could be reduced to < 0.7.16
Does Pulmonary Disease Alter the Toxic Effects of Hyperoxia?
A centuries-old debate is whether previously injured lungs are more or less vulnerable to hyperoxia. Several early O2 therapy practitioners believed injured lungs were less susceptible.1 Others (including notable researchers such as Lavoisier, Demarquay, Haldane, Comroe, and Tierney) believed hyperoxia in the presence of acute pulmonary injury may exacerbate inflammation,1,17,18 reasoning that “any disease that injures the alveolar wall may be adversely affected by concentrations of O2 that also injure the alveolar wall.”18 However, the pulmonary capillary endothelium appears to be the primary target cell initiating HALI (see HALI: Basic Cellular Pathways, below). Therefore, ALI-induced altered permeability pulmonary edema, which increases the alveolar-capillary O2 gradient, in theory might also afford some protection against secondary hyperoxic injury.19
In 1916, Karsner apparently was the first to provide some preliminary insight into this debate. He established that many seemingly healthy experimental animals in fact suffered from subclinical pulmonary disease.1 Yet in his experiments the toxic effects of hyperoxia in animals with subclinical pulmonary infection were indistinguishable from those he carefully screened prior to study.
Several experiments have explored how ALI might affect the subsequent development of HALI.3,15,18–22 Of particular interest were the initial studies of Ohlsson,3 who induced either moderate or severe ALI in rabbits by phosgene gas inhalation, and then were exposed to an FIO2 of 0.8 for 2 weeks. Whereas all control rabbits died from respiratory failure within 7 days, most lung-injured rabbits survived for approximately 2 weeks before developing overt signs of respiratory distress. In fact, moderately lung-injured rabbits survived longer (14–26 d) than severely lung-injured rabbits. However, blast-induced ALI seemingly had no effect on the development of HALI.3
Several studies used an oleic acid model of ALI to study prolonged exposure to an FIO2 of 1.0.15,19–21 Winter et al21 reported that prior injury in rabbits did not reduce the incidence of HALI, but it did significantly delay the onset of death, in a dose-dependent (ie, oleic acid) manner, particularly when hyperoxia was delayed by 24 hours after the onset of ALI. However, enhanced survival time was relatively modest (2.5–3.4 d), compared to the results of Ohlsson.3 Likewise, Smith and colleagues19 found a similar increase in the survival time of pre-injured rabbits, compared to control rabbits (6 vs 3.5 d) that was enhanced if exposure to hyperoxia was delayed by 36 hours (7 vs 3.5 d). In contrast, both Hakkinen et al20 (who studied mice exposed to an FIO2 of 0.8) and de los Santos et al15 (who studied baboons exposed to an FIO2 of 1.0) found that pre-injury with oleic acid did not alter the response to hyperoxia. In these studies there was no delay between inducement of ALI and exposure to hyperoxia. A possible explanation for these differences is that delayed exposure to hyperoxia following the inducement of ALI (a clinically irrelevant scenario) may have allowed enough time for alveolar type II cell proliferation to counter the toxic effects of O2.19
The modifying effects of acute pulmonary infection on subsequent hyperoxia are of particular interest. Tierney et al18 found that producing influenza pneumonia in both rats and mice 3 days prior to breathing an FIO2 of 1.0 resulted in decreased survival time (1–2 d), compared to either hyperoxia alone or with influenza at an FIO2 of 0.21. These findings support an earlier unpublished finding of influenza-infected mice exposed to hyperoxia, who had a 4-fold increase in mortality, compared to mice breathing room air.23
More recently, Thiel et al22 demonstrated in a polymicrobial infection model of ALI that hypoxia paradoxically stimulates a lung-protective mechanism by increasing the biologic effect of adenosine by up-regulating the adenosine A2A receptor. This effect then suppresses immune cell response to bacterial toxin-induced alveolar injury. In brief, mice exposed to an FIO2 of 1.0 had greater lung injury and died rapidly, compared to mice exposed to room air. Although exaggerated lung injury also was found at an FIO2 of 0.6, it did not enhance short-term mortality. Intratracheal injection of an adenosine A2A receptor agonist protected the lungs from hyperoxia.
In summary, data from most models of ALI do not provide strong, consistent support for the hypothesis that injury preceding exposure to hyperoxia affords protection. In most instances in which it did, the effect was partially dependent upon a substantial delay between the initial lung injury and exposure to hyperoxia: a scenario that has little clinical relevance. In fact, data from infectious models of ALI suggest that hyperoxia likely augments pulmonary injury.
Interactions Between HALI and Ventilator-Induced Lung Injury
In the past decade, several studies have examined the combined effects of hyperoxia and mechanical lung-stretch on the induction of ALI.24–30 Mechanical ventilation of rodents at a tidal volume (VT) of 20 mL/kg with an FIO2 of 1.0 caused significantly more pulmonary edema, compared to the same VT with an FIO2 of 0.21.24 Likewise, the combination of high VT and hyperoxia in other studies caused significantly greater reductions in lung compliance, increased alveolar-capillary membrane permeability, more severe pulmonary surfactant dysfunction,25 and increased expression of pro-inflammatory mediators.29,30 Most notably, altered pulmonary capillary permeability, neutrophil infiltration, and elevated pro-inflammatory mediator release were largely avoided when a lung-protective VT of 6 mL/kg was used in conjunction with hyperoxia.29 Another study found that rabbits ventilated with VT of 25 mL/kg and a sub-toxic FIO2 of 0.5 had significantly increased lung injury scores, compared to those ventilated with the same VT and an FIO2 of 0.21.26 The combination of high stretch with an FIO2 of 0.5 produced increased alveolar-capillary permeability and increased neutrophil concentration in alveolar fluid.26
Interestingly, pre-exposure to hyperoxia for 12 hours before initiating high-stretch ventilation (a common clinical scenario) produced severe diffuse interstitial edema, hemorrhage, and neutrophil infiltration within 4 hours.27,28 Yet initiating high stretch ventilation without prior hyperoxia, hyperoxia alone, or both strategies combined, all produced comparatively mild pulmonary edema and neutrophil infiltration. What is most striking about these studies is the relatively rapid induction of ALI, suggesting either an additive or synergistic effect of high VT and hyperoxia.
HALI and Pneumonia
Although pneumonia has been described in experimental HALI,1,4,5,7,31 it is unclear whether this represents secondary bacterial infection or simply a non-infectious, irritation-induced bronchopneumonia.31,32 Prolonged exposure to hyperoxia causes tracheobronchitis, absorption atelectasis, inhibition of mucociliary transport function, and decreased bacterial clearance, as well as functional impairment of alveolar macrophages.32–35 Collectively, these increase the risk of infectious pneumonia and may contribute to ALI. Minor incidences of bacterial pneumonia have been reported in baboons breathing an FIO2 of 1.0.15,31 In one human study of hyperoxia in 1939, the most severely affected subject required brief hospitalization for pneumonia.1 Pneumonia has also been reported as a clinical complication of HALI.16
Recent studies have reported that hyperoxia markedly increased the lethality of both Legionella pneumophila and Pseudomonas aeruginosa in mouse models of pneumonia-induced ALI.36,37 For example, inoculation of mice with P. aeruginosa followed by exposure to 60 hours of an FIO2 of 0.9 resulted in a 70% mortality.36 In contrast, there was no mortality in mice exposed to either hyperoxia or bacterial inoculation alone. Apparently, this combination accentuates alveolar cell death, leading to loss of barrier integrity and systemic dissemination of bacteria.36 Moreover, concern has been raised about the apparent association between ventilator-associated pneumonia and prolonged exposure to hyperoxia.38
In summary, hyperoxia disrupts normal pulmonary defense mechanisms that theoretically increase the risk of secondary bacterial pneumonia. Yet animal models of HALI have not reported high incidences of bacterial pneumonia as a complication, and these studies suggest that widespread bronchopneumonia observed in the laboratory setting is likely irritation-induced. However, more recent animal models of pneumonia-induced ALI suggest that, in the clinical setting, hyperoxia might potentiate morbidity.
Studies in Lower Primates: Speculation About Human Susceptibility to HALI
Poikilothermic animals, as previously mentioned, are immune to HALI, presumably due to an inherently lower metabolic rate.7 Because the inflammatory effects of O2 mainly have been described in small animals with relatively higher metabolic rates, generalizing these results to humans is problematic. Primate models may reflect better the importance of HALI for humans, as their genomic structure, underlying physiology, metabolism, and pattern of injury development are closer to humans than that of other mammals.7,39,40 These studies typically incorporated “Old World” monkeys who diverged genetically from our common ancestors approximately 25 million years ago.40 Experiments examining the pathophysiology of HALI in lower primates have provided mixed results, mostly in regards to mortality (Table 1).7,8,12–15,31,39,41,42
Lethality of Hyperoxia in Primate Studies
In terms of histopathology, most studies have reported the classic findings of an early stage characterized by acute exudative pulmonary edema,7,8,13,15,31,39,42 followed by a subacute fibroproliferative stage8,13–15,31,42 Depending upon the day of death or sacrifice, these changes sometimes were found together.15,31 Other less consistent findings included focal areas of hemorrhage,8,13 hyaline membranes,13,31,41 pulmonary capillary damage,14,39 late emphysematous changes,7,41,42 and bronchopneumonia.7,31 In general, the degree of damage matched the intensity of observed respiratory distress. Animals appearing relatively unaffected by hyperoxia generally had only modest histopathologic changes (eg, squirrel monkeys42 and many of the rhesus monkeys studied by Friedrich and Greyzell7).
The oldest and perhaps most positive study (ie, supporting the notion that humans are less vulnerable to HALI) was by Friedrich and Grayzel.7 They compared rhesus monkeys to rabbits breathing at an FIO2 of 0.9 over 20 days. Whereas all rabbits characteristically developed respiratory distress and died within 4 days, rhesus monkeys did not develop respiratory distress until day 5 and typically survived between 6 and 18 days.7 Likewise, when Robinson13 subjected rhesus monkeys to an FIO2 of 0.85–0.9 the mortality was 38%, with all deaths occurring between 12 and 16 days. However, monkeys subjected to an FIO2 of 1.0 had a mortality of 60%, with death occurring between 4 and 7 days.13
When analyzed collectively, these studies are difficult to interpret, given the variety of study objectives (eg, mortality was not necessarily the primary outcome), test protocols, study duration, small sample sizes, the sometimes vaguely reported results, and different species. Overall, these studies suggest that: baboons appear to be more susceptible to HALI than other monkey species; onset of toxicity and mortality appear lower than in non-primate species; and the average time to death among lower primates appears longer than other animals. However, these results must take into account that a considerable number of primates develop respiratory distress and die from respiratory failure as quickly as animals genetically more distant from humans. In addition, the duration of several study protocols was only between 4 and 8 days (see Table 1), so that the apparent mortality may be underestimated.
More meaningful results for humans would require studies of our closest relatives, the chimpanzees (who diverged from our common ancestor approximately 3 million years ago).43 However, we could not find any hyperoxia studies of higher primates in our literature search. In summary, some of the studies reviewed suggest humans might be less susceptible to HALI. Nonetheless, when these studies are evaluated collectively, they do not provide convincing evidence that our closer relatives necessarily have a higher tolerance for hyperoxia. Therefore, lower primate studies do not warrant confident speculation about the risk of hyperoxia to humans.
Evidence of HALI in Humans
Controlled Experiments
The study of hyperoxia in humans with normal lungs is greatly limited, as our knowledge necessarily is restricted to very early signs and symptoms. In the early to mid-20th century several small studies found that breathing at an FIO2 of 0.96–1.0 for 48 hours did not produce symptoms of toxicity in most men.1 When reported, the most common early symptoms were substernal pain (aggravated by deep inspiration), bronchial irritation, cough, sore throat, and nasal congestion.17 Substernal distress was absent in all subjects exposed to an FIO2 of ≤ 0.5, but was common when exposed to an FIO2 of 0.75.17 Other symptoms have included hyperventilation, dyspnea, fatigue, paresthesia, headache, nausea, and vomiting.1,5,44 In some subjects symptoms appeared within 4–6 hours, whereas others could tolerate up to 65 hours before the symptoms became oppressive. The maximal tolerance recorded for normal subjects exposed to an FIO2 of 1.0 was 110 hours.45
A consistent finding among normal subjects with prolonged exposure to an FIO2 of 1.0 was a fall in vital capacity. This was associated with either absorption atelectasis or a restrictive breathing pattern caused by substernal pain.5,17,44 The threshold for producing signs and symptoms of O2 toxicity in most humans appears to be 24 hours breathing an FIO2 of 0.75, whereas several days exposure to breathing an FIO2 of 0.55 or less does not appear to produce signs of toxicity.5,17
Clinical Hyperoxia and ARDS
It seems to me that one problem is determining which is cart and which is horse. Isn't it true that the patients who are sicker are the ones who have more pulmonary disease and are therefore more likely to be given a high concentration of O2 for longer time?
—Roman W DeSanctis MD46
Beginning in the 1950s, reports of HALI in humans began to appear in the medical literature as O2 therapy became more prevalent in clinical practice.47,48 However, it was only in the 1960s, with the establishment of the ICU and prolonged mechanical ventilation, as well as hyperbaric O2 therapy, that serious concern over clinical O2 toxicity arose in response to numerous case reports in both adults and neonates.16,23,49–67
The first clinical report of apparent HALI in adults was published in 1958.47 Other reports soon appeared associating O2 therapy with pulmonary hyaline membrane formation in adults who died of progressive respiratory failure.23,49–53 An early report by Cederberg50 in 1965 was influential in framing the initial theory regarding HALI, while also exemplifying the limitations of virtually all subsequent reports published in the 1960s and 1970s (Table 2). For example, at that time hyaline membrane formation was widely considered to be a hallmark of HALI. However, only a minority of subjects (19%) had that finding on autopsy. Interestingly, the propensity to develop pulmonary hyaline membranes during HALI is species-dependent,5 and the original study68 describing it in humans remarked that it had been a prominent finding among patients dying of viral pneumonia during the 1918 influenza pandemic (ie, ARDS).69
Clinical Case Studies Describing Potential Oxygen Toxicity in Adults
Half of the patients studied by Cederberg et al50 presumed HALI could be explained by other factors (eg, pulmonary infection, radiation therapy). In fact, many patients had risk factors for ALI (eg, aspiration, blood transfusions, sepsis, trauma, cardiopulmonary bypass), a fact not appreciated in the 1960s. Furthermore, the highest FIO2 (mean of 0.53) was too low and the duration of exposure (mean of 22 h) was too brief to produce HALI based on previous animal and human studies. This exaggerated emphasis on HALI was exemplified by Sevitt,66 who suggested that breathing an FIO2 of 0.4 for several days was potentially dangerous!
In February 1967, Northway et al52 and Nash et al53 published their seminal reports linking mechanical ventilation with high FIO2 to progressive respiratory failure and death in neonates and adults, respectively. This brought the issue of HALI to the forefront of acute care practice and stimulated a groundswell of subsequent case series reports.16,54–67 Nash et al53 found the greatest degree of pulmonary pathology in those patients mechanically ventilated for greater than 10 days and with an FIO2 > 0.9. The lungs of these patients were nearly identical to the previous descriptions from animal studies of HALI, including a characteristic early “exudative phase that progresses … to a proliferative or fibrotic phase.”53 Nash et al cautioned that they had not established a cause-and-effect relationship, and that “physical factors such as the pattern of ventilation and high airway pressure” had to be considered.53 Notwithstanding, he believed the term “respirator lung syndrome” was a misnomer and strongly implied that prolonged exposure to a high FIO2 was the primary culprit.
Likewise, Northway et al52 reported the use of peak airway pressures up to 40 cm H2O and prolonged (6 d) exposure to an FIO2 ≥ 0.8 in neonates. However, in contrast to Nash et al's description of HALI as “a gradual progressive deterioration of pulmonary function apparently unrelated to the disease that necessitated respiratory assistance,”53 Northway et al described bronchopulmonary dysplasia as emerging from an initial phase (2–3 d) of severe injury “indistinguishable from any severe case of the respiratory distress syndrome.”52
Interestingly, the term “respirator lung” was first coined by Linton and colleagues,70 who briefly described the phenomenon among 200 mechanically ventilated patients from 1960 to 1965. Accordingly, “respirator lung” usually began with bronchospasm and decreased lung compliance. Initially it could be treated effectively with bronchodilators and “higher inflation pressures which improved ventilation,” but later these were progressively less effective.70 Chest radiographs in these patients showed “widespread bronchopneumonia”; “finally, pulmonary ventilation became impossible, very high inflation pressures cannot force enough O2 into the lungs to maintain adequate oxygenation.”70 At autopsy the lungs had “a consistency of liver, the alveoli being completely filled with inflammatory exudate.”70 Most patients had positive sputum cultures for organisms commonly found in ventilator-associated pneumonia (eg, pseudomonas). Beginning in 1964 the hospital modified their infection control policy for mechanically ventilated patients, which reportedly eliminated the syndrome.
In a subsequent laboratory study, Nash et al71 reported that only spontaneously breathing or mechanically ventilated goats with an FIO2 of 1.0 developed HALI. However, these animals were ventilated at a very low airway pressure (13 cm H2O) that was unlikely to produce lung injury. A similar study of lambs exposed to an FIO2 of 0.8–1.0 also failed to demonstrate a relationship between mechanical ventilation and pulmonary lesions associated with HALI.72 However, specific information regarding VT and airway pressures was lacking. Both studies failed to consider a prior investigation by Greenfield et al,73 wherein mechanically ventilated dogs subjected to “over-inflation” for 24 hours caused either depletion or alteration in pulmonary surfactant and atelectasis. These authors reasoned that prolonged mechanical ventilation with a sufficiently large VT could interfere with normal pulmonary surfactant function that theoretically could lead to hyaline membrane formation.
Tidal Volume Practice During Mechanical Ventilation in the 1960s
Any analysis of HALI must consider the state of mechanical ventilation practices in the 1960s, as well as the profound limitations in the available technology. A case study46 published along with the Nash et al53 paper illustrated the difficulties in assessing other contributing factors in cases of suspected “traumatic oxygen alveolopathy” (eg, chest trauma, septicemia, pneumonia, fat embolism). The patient had been ventilated with a VT of 800 mL despite a pulmonary compliance of 16 cm H2O/mL,46 which may have produced alveolar pressures approaching 50 cm H2O. An equally enlightening study, also from the same institution as the Nash et al study, revealed that during the early to mid-1960s at Massachusetts General Hospital patients commonly were ventilated at 850 ± 220 mL, or 13.2 ± 4.0 mL/kg measured body weight.74
In another case of presumed HALI, Sevitt66 also reported an early (36 h) rise in peak airway pressures to 50 cm H2O, which subsequently resulted in a tension pneumothorax. Taken together, these reports suggest that ALI and ventilator-induced lung injury either coexisted with or were mistaken for HALI. In fact, the recent evidence24–30 on the additive effects of hyperoxia and high stretch mechanical ventilation strongly implicates both practices. Similar findings have been reported in neonatal pigs subjected to hyperoxia and hyperventilation,75 with similar conclusion apparently reached in regards to risk factors for bronchopulmonary dysplasia.76
FIO2 Control During Mechanical Ventilation in the 1960s
Another important problem was the poor control over FIO2 with pressure-cycled ventilators commonly used in the early 1960s (eg, Bird Mark and the Bennett PR series).77,78 These ventilators controlled FIO2 through a primitive Venturi air-entrainment system that essentially was nonfunctional when encountering modest airway pressures. For example, when the Bird Mark 7 operated at a peak airway pressure of only 20 cm H2O, the FIO2 was > 0.9.77 Another study found that the FIO2 approached 1.0 when these ventilators reached an airway pressure of only 12 cm H2O.78 Excessive FIO2 delivery despite engagement of the air-O2 mix function also was documented in several case reports of HALI.16,59 Therefore, it is quite possible that the pervasive use of pressure-cycled ventilators in the 1960s may have caused numerous cases of HALI. Unfortunately, we will never be certain of the extent to which this may have occurred.
Other Clinical Factors Influencing HALI
Finally, as Nash et al observed, “a multitude of biochemical changes associated with both the disease process and the treatment” may obscure the attribution of lung inflammation to HALI.53 HALI is strongly influenced by factors such as hormones, drugs, and morbid conditions. For example, fever, hypercarbia, insulin, epinephrine, and norepinephrine enhance toxicity, whereas hypothermia, barbiturates, antihistamines, sodium bicarbonate, and tris-buffer ameliorate it.5,79 Obviously, these co-factors may influence the severity of HALI in ways that cannot be determined clinically.
Is HALI a Clinically Relevant Problem in the 21st Century?
Compared to the 1960s, current attitudes regarding the risks associated with hyperoxia seem relatively relaxed. What transpired clinically that might account for this impression? Six months after the publication of the Nash et al study,53 the theory of HALI was greatly overshadowed when Ashbaugh et al80 published the landmark description of ARDS and introduced the technique of using PEEP to treat refractory hypoxemia. Both the clinical and pathologic findings described by Ashbaugh et al were virtually the same as HALI. However, hyperoxia was categorically rejected as a contributing factor. Instead it was argued that the observed pulmonary inflammation was a stereotypical pathologic response to multiple initiating factors, such as viral pneumonia, acute pancreatitis, drug overdose, and trauma. This case series crystallized disparate historical accounts of acute respiratory failure in adults (eg, congestive atelectasis, wet lung, shock lung) and revolutionized its treatment with the introduction of PEEP. Subsequently, numerous reports elaborated on the etiology and pathophysiology of ARDS, which shifted the focus away from HALI.81–84
Other contributing factors in the early 1970s included the evolution of mechanical ventilation that shifted practice away from pressure-cycled to volume-cycled ventilators, with precise control of FIO2. In addition, the advent of hospital-wide bulk O2 and compressed air availability facilitated more precise FIO2 delivery and eliminated the limitations from reliance upon tank O2.54
Probably the most important factor was the advent of PEEP. It became readily apparent that moderate to moderately high levels of PEEP allowed most patients with respiratory failure to achieve adequate arterial oxygenation at a relatively non-toxic FIO2 of ≤ 0.6. This strategy was elegantly captured in an influential editorial by Albert, who proposed a “least PEEP” strategy, whereby clinicians should target the lowest level of PEEP that secures adequate arterial oxygenation at a relatively safe FIO2 (ie, 0.6–0.7).85 Because ARDS represents one of the greatest challenges in managing hypoxemia, the findings of recent randomized controlled trials of early ALI/ARDS is illuminating. These trials reported that during the first 3 study days, mean FIO2 requirements were at or below clinically accepted toxic thresholds and were achieved with moderate or moderately high levels of PEEP (Table 3).86–91 Furthermore, as recent animal studies24–30 suggest, lung-protective ventilation may substantially reduce additional risks from hyperoxia. However, as with the current debate over the use of high PEEP and other ancillary therapies for improving oxygenation (eg, prone positioning, recruitment maneuvers, inhaled prostacyclin), there likely exists a subset of patients with severe ARDS in whom HALI is a legitimate concern, thus justifying the incorporation of these therapies.
FIO2 and PEEP Requirements of Patients With Acute Lung Injury Enrolled Into Randomized Controlled Trials of Mechanical Ventilation
Long-Term O2 Therapy and the Risk of Chronic Pulmonary Toxicity
An intriguing issue that cannot be tested pragmatically in animal models is whether or not prolonged exposure (ie, years) to “non-toxic” levels of O2 might cause chronic lung inflammation. Petty et al92 reported a case-series of 14 patients with severe COPD who were treated with low-flow nasal O2 therapy (estimated FIO2 of 0.22–0.27) for an average of 27 months prior to death. Approximately half of these patients had classic findings of capillary proliferation, interstitial fibrosis, epithelial hyperplasia, and, in 2 cases, acute exudation on autopsy exam. Aside from this study, very little evidence is available regarding the possibility of clinically relevant O2 toxicity in patients with COPD or other chronic cardiopulmonary diseases requiring long-term, low FIO2 therapy. More recently, evidence of “oxidative stress” in patients with COPD receiving short-term (18–48 h) supplemental O2 of 2 L/min has been reported.93 Supportive evidence of low-FIO2-induced toxicity in animal models comes from dogs exposed to an FIO2 of 0.33 for 8 months, who were found to have a 54% increase in alveolar-capillary thickness, with associated diffusion defect.5
However, environmental pollution produces numerous respirable oxidants to which patients with chronic pulmonary disease are particularly susceptible.9 The injurious effects include both oxidative lung injury as well as bacterial and viral infections, the susceptibility to which is highly individual.9 Therefore, among out-patients with chronic cardiopulmonary diseases, attributing lung injury primarily to long-term O2 therapy is problematic. Regardless, as Petty et al noted, the benefits of low-flow O2 therapy far outweigh any potential toxic risks.92
Overview of the Molecular Biology of HALI
The paradox of O2 is that it is both necessary for aerobic cellular life while simultaneously destroying it through the production of free radicals (molecules containing unpaired electrons) known as reactive oxygen species (ROS).48 Major advances in our understanding of HALI began only in 1954, when the hypothesis was advanced that abnormal ROS generation was responsible for both O2 and radiation poisoning.48 Hyperoxia causes initial cellular damage from the production of ROS, through the law of mass action, whereby the production of ROS is directly proportional to tissue PO2, and inversely related to electron flow in the cytochrome chain.94,95 Therefore, both increased tissue PO2 and related impaired cytochrome function (eg, cytochrome c depletion) generate excessive amounts of ROS. This in turn causes damage to macromolecules, leading to irreversible cellular dysfunction and death. Pulmonary injury is compounded through secondary ROS production generated by an immune response to the initial injury.
Basic Mechanisms of ROS Production and Cellular Damage
Aerobic metabolism involves the reduction of molecular O2 to form adenosine triphosphate and water. Four consecutive one-electron steps are involved in this process, and ROS are produced as intermediate metabolites.95,96 These include the pivotal and highly reactive superoxide anion (O2.–) hydrogen peroxide (H2O2), hydroxyl radical (OH–), and peroxynitrite anion (ONOO–).38,94–99 In the initial phase of HALI the most important source of ROS generation is the mitochondria (Fig. 1).95,99 During oxidative phosphorylation, energy transformation occurs at 3 protein complexes on the inner membrane of the mitochondria known as complex I, III, and IV.38,95,96 Normally, 1–2% of O2 molecules undergo “electron leakage” at complex I and III, creating a small steady state of superoxide anions.96

Mechanisms governing the initial burst in reactive oxygen species (ROS) in the primary target cell: the pulmonary capillary endothelium. 1: ROS generation is proportional to the PO2 during hyperoxia. 2: O2 molecules are reduced to form ROS by nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidase (NOX) at the plasma membrane. 3: Damage to the plasma membrane lipid bilayer by ROS also generates additional ROS. 4: The primary sources of ROS production occur inside the mitochondria. Inadvertent electron leakage normally occurs at intermediate steps of energy transformation on the cytochrome chain (protein complexes I and III), but increases proportionally with the intensity of hyperoxia. Additional sources of ROS are generated from the interaction of O2 molecules with numerous mitochondrial enzymes, including cyclooxygenases, peroxidases, lipooxygenase, and cytochrome P450. 5: As the endothelium is a rich source of nitric oxide (NO), reactions with O2 molecules and superoxide anion produces nitrogen dioxide (NO2), a reactive nitrogen species, and peroxynitrite anion (ONOO–), respectively. Peroxynitrite anion reacts with carbon dioxide to form additional NO2.
Mitochondrial ROS generation begins with O2.–, which undergoes dismutation to form H2O2; ionized iron (Fe2+) reacts with H2O2 to form highly reactive OH–.95,96,99,100 In the presence of nitric oxide, O2 forms radical nitrogen species, whereas O2.– forms ONOO–, which reacts with carbon dioxide to form additional radical nitrogen species.9 Other mitochondrial enzymatic reactions normally generate ROS, including cyclooxygenases, lipooxygenases, peroxidases, and cytochrome-dependent oxygenases.9,95,97 Normally ROS are balanced by cellular antioxidant defense mechanisms (see below).9,38,95,96,99 However, in the presence of hyperoxia mitochondrial ROS production increases linearly with intracellular PO2, thus overwhelming antioxidant defense mechanisms and further exacerbating ROS production.9,38,95,96 An interesting finding for clinicians is that mitochondrial H2O2 generation is biphasic, increasing at a faster rate when FIO2 is > 0.6.95
Hyperoxia triggers a secondary inflammatory response that generates ROS from activated macrophages, platelets, and neutrophils (Fig. 2).97,99,100 The intensity of this response is species-dependent,99 thus explaining some of the variable susceptibility of different animals to HALI. There also exists another important extracellular source of ROS. This involves the nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidase (NOX) enzyme family that utilizes NADPH to reduce O2 into O2.–.38 Hyperoxia also enhances ROS production by this pathway, in direct proportion to extracellular PO2.
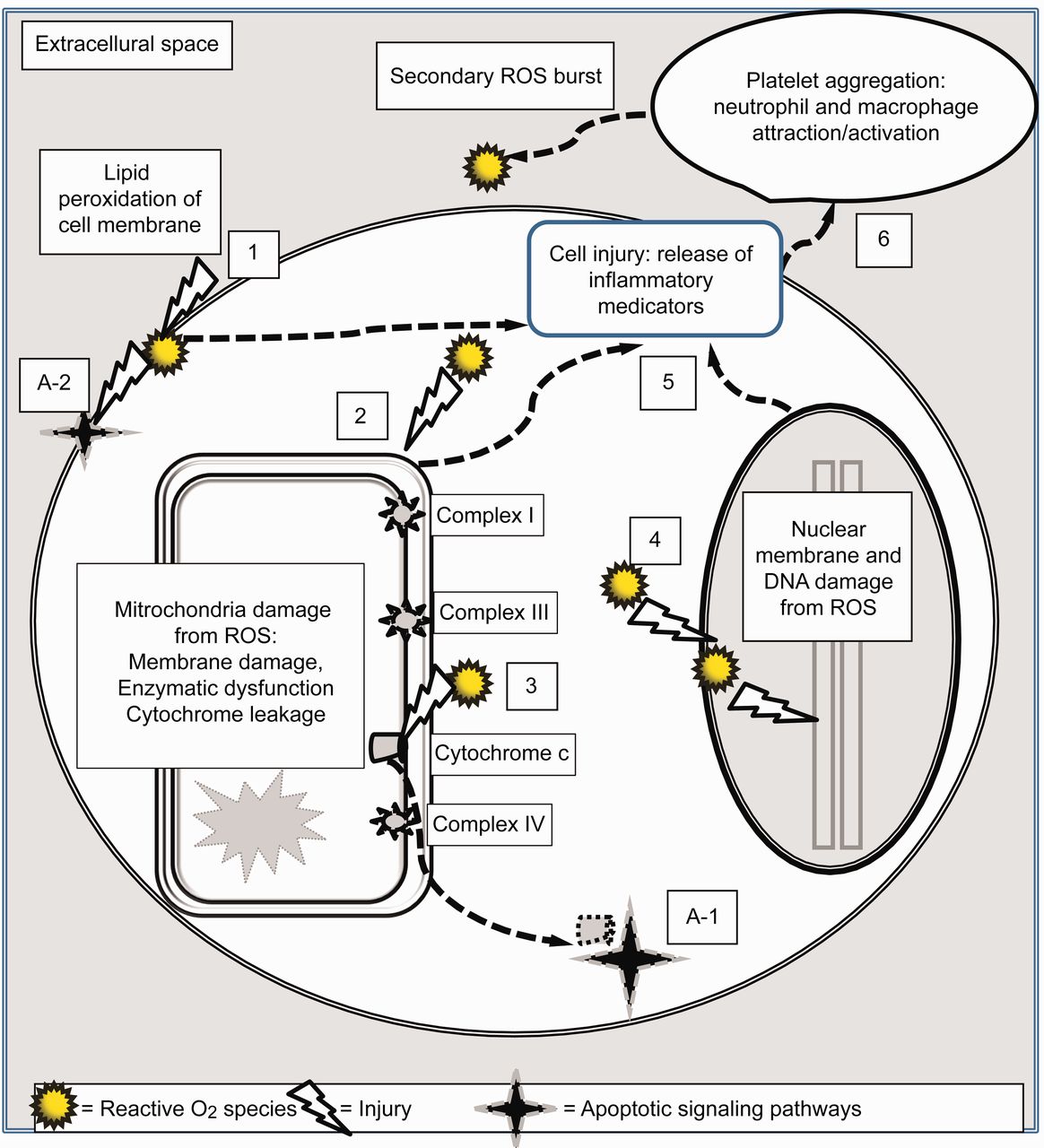
Mechanisms governing the secondary burst of reactive oxygen species (ROS) and basic pathways of cell death from hyperoxia. 1: Loss of plasma membrane integrity from lipid peroxidation by ROS. 2: ROS damage to the mitochondria membranes and deactivation of enzyme systems and cytochrome chain. 3: This results in the release of cytochrome c into the cytoplasm. 4: ROS damage to the nuclear membrane and fragmentation of DNA. 5: Evolving cell trauma from steps 1, 2, and 4 trigger the production and release of pro-inflammatory cytokines and chemokines into the extracellular space and circulation. 6: This attracts and activates platelets, neutrophils, and macrophages, resulting in a secondary burst of ROS from these inflammatory cells. Direct cell trauma results in necrosis or “unplanned” cell death. In addition, the release of cytochrome c into the cytoplasm (A-1) and damage to the plasma membrane (A-2) trigger other cellular processes that instruct the cell to essentially “commit suicide” through the process of apoptosis (programmed cell death).
ROS readily capture electrons from surrounding proteins and lipids, causing cell membrane rupture, enzymatic dysfunction, mitochondria, and DNA damage that ultimately results in cell death (see Fig. 2).9,38,94,97–99 For example, ROS interacts with the lipid component of cell membranes (“lipid peroxidation”), causing a complex series of reactions that not only break down the lipid bilayer, but generate additional ROS.97,98 ROS also disrupt cellular mechanisms, both by activating and deactivating specific proteins.97 A particularly important consequence is the inactivation of several antioxidant enzyme systems by ROS, thus enhancing tissue injury by weakening cellular defense mechanisms.96,97
Antioxidant Defense Mechanisms
Through evolution, all cells have developed elaborate defense mechanisms against ROS,94,97–99 and the term “oxidative stress” describes all destructive processes emanating from any imbalance between ROS and anti-oxidant defense mechanisms.95 In mammalian cells the main antioxidant enzyme system is superoxide dismutase (SOD), which consists of intracellular and extracellular copper-zinc SOD, and manganese SOD (found almost exclusively in the mitochondria) (Fig. 3).95 Adaptation to hyperoxia partly involves enhanced activity of both copper-zinc and manganese SOD.11 Other important antioxidant enzymes include catalase and glutathione peroxidase, which are complemented by a number of other non-enzymatic factors located in both the intracellular and extracellular compartments.94 These systems work in concert to reduce ROS to water.

Superoxide dismutase (SOD) is the primary anti-oxidant defense system operating at the cell surface, inside the cytoplasm, and inside the mitochondria. Outside of the mitochondria this involves copper-zinc form (Cu-Zn) SOD, whereas inside the mitochondria it is manganese (Mn) SOD. Donated electrons (E*–) reduce O2 molecules to form the highly reactive superoxide anion (O2*–), which SOD converts to hydrogen peroxide (H2O2). Through a series of secondary reactions involving glutathione, catalase, N-acetylcysteine, and other agents, H2O2 is converted to H2O.
In the first step, SOD converts the highly reactive O2.– into H2O2. Then catalase and glutathione peroxidase, as well as N-acetylcysteine, reduce H2O2 to water through a series of secondary reactions.97,100 Other non-enzymatic antioxidants include vitamins C and E, as well as β-carotene and uric acid.94 Therefore, the ability of any animal to survive HALI depends upon a balance between the magnitude and duration of excessive ROS exposure and the ability to marshal sufficient anti-oxidative as well as tissue-repair mechanisms.
In addition, adaptive mechanisms may vary by cell type, so that certain tissues may be more or less susceptible to oxidative damage.97 These adaptive mechanisms, in turn, are dependent upon the ability of cells to up-regulate gene expression that controls antioxidant production. This likely explains the variable susceptibility to HALI reported among different animal species, subspecies, and individuals. It also should caution clinicians against complacency toward allowing the prolonged exposure of patients to toxic levels of FIO2, as individual susceptibility to HALI currently is unknowable.
HALI: Basic Cellular Pathways
HALI causes alveolar cell death through vastly complex cellular and molecular pathways. Although hyperoxia damages the alveolar epithelium as well as macrophages and vascular endothelium, these may occur through distinct pathways.99 A consistent finding in animal models of HALI is the particular susceptibility of the vascular endothelium to hyperoxia,1,3,8,11,14,39,101–105 an observation that has been made in humans with suspected HALI.47,106 Over 70 years ago it was proposed that HALI “might be explained by a hypothesis of deranged vascular physiology.”1 Capillary endothelial damage is the precipitating step in HALI.3 It is more extensive and typically occurs earlier (between 40–60 h) than damage to the alveolar epithelium.11,39,102 Initial endothelial damage causes platelet aggregation occurring prior to neutrophil infiltration, both of which are important sources of ROS generation resulting in secondary injury.107
The pulmonary capillary endothelium's susceptibility to HALI may be related to its high metabolic activity from regulating numerous vasoactive and fibrinolytic substances that pass through the pulmonary circulation (eg, serotonin, norepinephrine, bradykinin, angiotensin, prostaglandins, tissue plasminogen activator).105,108,109 Harabin et al demonstrated that prolonged exposure to an FIO2 of 1.0 decreases endothelial metabolism by 50% in dogs.105 This accounts for pulmonary dysregulation of circulating vasoactive substances during hyperoxia110 and partially explains subsequent pre-terminal hemodynamic instability.105 Physiologic states associated with increased metabolism are known to enhance HALI,79 probably because of elevated oxidative phosphorylation and increased mitochondrial ROS production.
Also, the vascular endothelium is a rich source of nitric oxide, which, as noted above, may potentiate cellular injury by secondary generation of both ROS and radical nitrogen species.9 In fact, this may explain the puzzling findings of Ohlsson,3 who reported that the combination of 3% inspired carbon dioxide to an FIO2 of 0.8–0.9 greatly accelerated pulmonary damage and markedly decreased survival time. Therefore, the pulmonary capillary endothelium may be particularly susceptible to hyperoxia. In fact, high stretch mechanical ventilation increases endothelial metabolism, which theoretically may exacerbate HALI.109 Alternatively, the endothelium may not possess the same antioxidant-generating capabilities as other alveolar cell types.
Cytokines and HALI.
The cellular pathways of HALI involve the interactions of “signaling molecules” such as cytokines (peptides that regulate inflammation and tissue repair),101 tissue growth factors such as angiopoietin-2 (destabilizes blood vessels, enhances capillary leak),2 and nitric oxide.111 In brief, hyperoxia causes initial damage to the alveolar endothelium and epithelium, inducing them to release interleukin-1. This initiates the production and release of other cytokines, including interleukin-6, and interleukin-8. In turn, these cytokines stimulate the release of numerous other molecules that attract and/or activate neutrophils, macrophages, and other inflammatory cells, causing increased vascular permeability and secondary ROS production (see Fig. 2).101,111–113 Hyperoxia also stimulates the production of vascular endothelial growth factor, which has both injury-enhancing and lung-protective properties.101 It is most likely responsible for pulmonary capillary proliferation during hyperoxia, first described by Pratt.47
Anti-inflammatory cytokines such as interleukins 10, 11, and 13, and certain growth factors, also are up-regulated in response to hyperoxia.101 In particular, interleukin-11 appears to limit lipid peroxidation and alveolar capillary permeability, as well as DNA damage.101 Keratinocyte growth factor may induce alveolar type II cell hyperplasia that characterizes the adaptive response to chronic hyperoxia.101 Additionally, this growth factor ameliorates DNA damage and may protect both the alveolar epithelium and endothelium.101 These cytokines exert a protective effect, both by stimulating cellular antioxidant defense mechanisms and by attenuating “cell death signaling pathways.”35
Necrosis Versus Apoptosis: The Complexity of Cell Death During Hyperoxia
A fundamental pathway in HALI is the triggering of apoptosis or “programmed cell death” via signaling molecules that essentially instruct cells to “commit suicide.” In contrast, necrosis or “unscheduled cell death” occurs through trauma such as lipid peroxidation of the cell membrane, enzymatic dysfunction, and DNA fragmentation, as typically occurs in HALI. Microscopically, apoptosis is characterized by cell shrinkage and “blebbing” of an intact cell membrane, whereas necrosis features membrane lysis and cellular swelling.112 Both processes overlap during HALI through highly complex and redundant pathways that ultimately lead to cell death.35,94,99,100,104,114
Initiation of apoptosis occurs both through extrinsic and intrinsic pathways (see Fig. 2). Extrinsic pathways involve the triggering of cell surface receptors, known as a “death-inducing signal complex,” that activate proteases (enzymes that initiate protein catabolism) such as caspace-8.38 Intrinsic pathways are believed to be more important during HALI.99 It is initiated by mitochondrial membrane damage from ROS and the release of cytochrome c into the cytoplasm, which in turn activates caspace-9.35 Adding to this complexity, other intracellular molecules, such as toll-like receptor 4 (a protein integral to immunity), appear to counter apoptosis and promote survival in experimental animals during hyperoxia.35
Summary
Prolonged breathing of extremely high FIO2 (≥ 0.9) uniformly causes severe HALI in virtually all animal species, and, without a reduction of FIO2, is usually fatal. However, the lethal threshold for humans exposed to prolonged hyperoxia is unknown and cannot be speculated upon with any confidence based upon lower primate studies or clinical reports. Interestingly, recent speculation posits that a lethal dose for the “average human” might require extended exposure in a slightly hyperbaric range (1.1–1.2 atmospheres).115 However, the authors emphasize that the wide dose range for O2 lethality demonstrated in animal studies also suggests that a segment of the population very likely would remain vulnerable at an FIO2 approaching 1.0.
The severity of HALI is directly proportional both to the PO2 (particularly above 450 mm Hg, or an FIO2 of 0.6) and the duration of exposure. Hyperoxia produces high amounts of ROS that overwhelm natural anti-oxidant defenses. ROS destroy cells both directly, through damaging cellular structures, and indirectly, through instructing cells to “commit suicide.” Genetic predisposition has been shown to play an important role in HALI among animals, and some genetics-based epidemiologic research suggests that this may be true for humans as well. Such susceptibility probably reflects the response to injury through a balance between complex pro- and anti-inflammatory cellular mechanisms.
The evidence presented in this review suggests the following. First, in the absence of high-stretch mechanical ventilation, the risk of HALI is minimal when the FIO2 is ≤ 0.6; the risk probably begins when the FIO2 exceeds 0.7, and likely becomes progressively more problematic as the FIO2 exceeds 0.8 for an extended period of time. Second, the primitive control of FIO2 during mechanical ventilation in the 1960s may have resulted in numerous incidents of HALI, which was potentiated by the concurrent use of high VT ventilation. However, the attribution of progressive respiratory failure and death to hyperoxia in the 1960s is tenuous, given the fact that both ARDS and ventilator-induced lung injury had not yet been recognized. Third, the advent of PEEP and precise control over FIO2, as well as lung-protective ventilation and adjunctive therapies for severe hypoxemia, has substantially reduced the risk of HALI for the vast majority of patients requiring mechanical ventilation in the 21st century. However, the subset of patients with very severe ARDS requiring prolonged hyperoxic therapy may be at increased risk of HALI. This may be magnified in those with viral pneumonia as well as those in hyper-metabolic states such as sepsis or trauma. Thus, the use of adjunctive therapies that improve pulmonary O2 transfer is indicated in these patients.
Discussion
Heffner:
With the potential for O2-induced injury from hyperoxia, it seems that differences in species had previously been explained by differences in glutathione reductase content and some differences between neonate and adult animal vulnerability. Is there anything new regarding the role of antioxidants in protecting against hyperoxic lung injury? And what are the implications inherent in the occurrence of hyperoxic lung injury in the setting of amiodarone or bleomycin therapy, which increase the risk for patients, like a one-two punch?
Kallet:
The diagrams I did for this paper are very basic, just to give an idea of cellular signaling pathways and interactions. In reality the complexity at the cellular and molecular level is overwhelming and fascinating. There is concern with drugs such as amiodarone, bleomycin, and adriamycin, which basically increase the production of radical O2 species, and which are potentiated by hyperoxia.
There has been some encouraging animal data on co-administering drugs like resveratrol with bleomycin to ameliorate oxidative damage.1 A recent review on the pulmonary toxicity of amiodarone mentioned that treatment with vitamin E had been effective in some patients with pulmonary fibrosis, but it's not been recommended as part of standard therapy, and it's unknown if it would have any effect in patients with more severe amiodarone-induced toxicity leading to ARDS.2
Perfenidone and N-acetylcysteine have been effective in the treatment of idiopathic pulmonary fibrosis.3 N-acetylcysteine supplements also have been used effectively in patients with COPD who had what they called oxidative stress (increased cytokine production) from low-flow O2 therapy.4 And this occurs just after a few hours of exposure. They observed, both in the blood cells and plasma samples, that there was oxidative stress they could treat with N-acetylcysteine.
However, despite those encouraging results, it is interesting that all the large studies of antioxidant therapy in ARDS that I am aware of have been very discouraging. That suggests to me that, in these acute inflammatory processes, by the time we identify and begin antioxidant therapy, the process of oxidative damage is so revved-up and pervasive that we can't effectively intervene with exogenous antioxidants.
Criner:
In a Pulmonary Perspective paper,5 Rick Albert said that ALI/ARDS is largely an iatrogenic disease—that atelectasis is the primary cause, and that the inflammatory milieu that occurs with it is actually a consequence of the syndrome developing, which is iatrogenic, from the way we ventilate patients and surfactant deficiencies that occur with how we position patients, et cetera. One thing he didn't really talk about is the effects of supplemental O2 causing surfactant dysfunction and how that could lead to atelectasis. What do you think about that: that a predominant complication of O2 therapy is contributing more to surfactant dysfunction?
Kallet:
It obviously plays a role, but I think it's way too complicated to lay it all on that. I don't buy into ARDS being primarily iatrogenic: I really don't! It could in some ways harken back to the question of should we give O2 to people with lung inflammation? And it's kind of another way of saying that. It does get to the idea that, if you have someone with hypoxemia, you're going to give them O2 to get their saturation up. It's the lung taking a hit for the team. Someone with massive chest and abdominal trauma is going to get ARDS regardless of what we do! That means it's the degree of insult, and I think it's a little too simplistic to lay it at the feet of atelectrauma or high-stretch ventilation.
There's a reason these people are on the ventilator, and most patients with ARDS are intubated for pneumonia or secondary trauma or aspiration or pancreatitis. The inflammatory response is already engaged. I do think it's appropriate to say the severity of the response is somewhat under our control by how we manage the patients, but I think our hands are tied to some degree.
Branson:
Since ARDS is primarily a pulmonary capillary leak syndrome, isn't the presence of the fluid in the lung a powerful antioxidant? And isn't that one of the reasons that having lung injury might be protective against oxygen toxicity? There are a lot of patients who are on 40% O2 and have a PO2 of > 150 mm Hg, and, because the classic thinking is that FIO2 less than 60% is not toxic, I'm not going to turn the FIO2 down. I don't know if maybe that creates some kind of non-pulmonary O2 toxicity.
Kallet:
The way you phrased that, I thought you meant whether there was any antioxidant effect from alveolar capillary leak. I guess a way of rephrasing your question would be to ask if the diffusion barrier resulting from alveolar edema produces a PaO2 of 60 mm Hg, despite a “theoretical” local alveolar PO2 of 600 mm Hg, wouldn't the alveolar tissue be protected? That would assume that the PaO2 is equivalent to the pulmonary capillary PO2. But in the presence of a large intrapulmonary shunt that might be misleading. It might be similar to ventilator-induced lung injury in that relatively undamaged pulmonary tissue would more readily diffuse O2 across, causing oxidative damage to epithelium and particularly the endothelium.
In reading through the literature, it seems that it's more cellular signaling that stimulates certain genes to rev up glutathione production, and you get pretty quickly overwhelmed by the number of factors and molecules. It's this dialectical balance between a myriad of different pro- and anti-inflammatory triggers that affects this. It still appears that a tissue PO2 of greater than 400 mm Hg is when the antioxidant defense mechanisms get overwhelmed. But then again, it isn't all that clear to me whether all of the body's tissue beds have the same level of antioxidant defenses that the lungs do. To be on the safe side, under most circumstances we shouldn't let patients breathe at a higher than necessary FIO2 for an extended period of time, probably measured in hours.
Branson:
I hate to steal this from Dave Pierson, because I think he's the first person who said it, but oxygen toxicity is like Bigfoot: everybody's heard about it, but nobody's ever seen it.
Kallet:
One of the things I think I illustrated is that you're almost never going to be able to definitively lay it down on that. It's too complicated.
Footnotes
- Correspondence: Richard H Kallet MSc RRT FAARC, Respiratory Care Services, San Francisco General Hospital, NH:GA-2, 1001 Potrero Avenue, San Francisco CA 94110. E-mail: rich.kallet{at}ucsf.edu.
Mr Kallet presented a version of this paper at the 50th Respiratory Care Journal Conference, “Oxygen,” held April 13–14, 2012, in San Francisco, California.
The authors have disclosed no conflicts of interest.
- Copyright © 2013 by Daedalus Enterprises Inc.
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.
- 34.
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.
- 52.↵
- 53.↵
- 54.↵
- 55.
- 56.
- 57.
- 58.
- 59.↵
- 60.
- 61.
- 62.
- 63.
- 64.
- 65.
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.
- 83.
- 84.↵
- 85.↵
- 86.↵
- 87.
- 88.
- 89.
- 90.
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.
- 104.↵
- 105.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 113.↵
- 114.↵
- 115.↵





{kind=link}
{kind=link}
{kind=link}