Article Text

Abstract
This report presents a case of eosinophilic angiocentric fibrosis in a man with Wegener's granulomatosis, the first report of a possible association between the two conditions. This association suggests a possible mechanism for its pathogenesis.
- Wegener's granulomatosis
- eosinophilic angiocentric fibrosis
- nose
Statistics from Altmetric.com
Eosinophilic angiocentric fibrosis (EAF) is a rare inflammatory lesion of the nose and upper airways, first described in 1985.1 Its aetiology is unknown, but it has been reported associated with granuloma faciale and atopy.1 Most patients also have had surgery in the area before developing the distinctive perivascular fibrosis of the lesion.1–3 We present the first report of EAF in association with Wegener's granulomatosis (WG). This association raises questions regarding the aetiology and pathogenesis of this lesion.
Clinical details
A 42 year old man with WG presented to our ear, nose, and throat services with nasal obstruction and postnasal drip. He had a history of mild asthma since childhood, for which he took inhaled salbutamol as required. He had presented five years previously with nasal obstruction, when he underwent intranasal antrostomies and submucosal resection of his nasal septum, the histopathology of which first suggested WG (see below). The clinical findings of scleritis, flitting arthralgia, one documented episode of haematuria, and a raised creatinine of 117 μmol/litre (normal, < 110 μmol/litre) supported this diagnosis. Positive cytoplasmic pattern antineutrophil cytoplasmic antibodies (C-ANCA), which rose from a titre of 1/40 to 1/640 during his illness, settling after treatment, and raised antiproteinase 3 (PR-3) antibodies (5 U/ml; normal, 0–2 U/ml) confirmed the diagnosis. Chest radiographs, however, were normal and further tissue diagnosis was not sought.
He received a full course of immunosuppressive treatment for WG, following which he made a symptomatic recovery, also reflected in normalisation of his biochemical markers.
On his present admission his right nares was obstructed, with nasal septal deviation to the right. Further clinical examination was unremarkable. C-ANCA positivity remained low (1/20), with C reactive protein and creatinine within normal limits. He underwent an elective operation for division of adhesions without event.
Histopathology
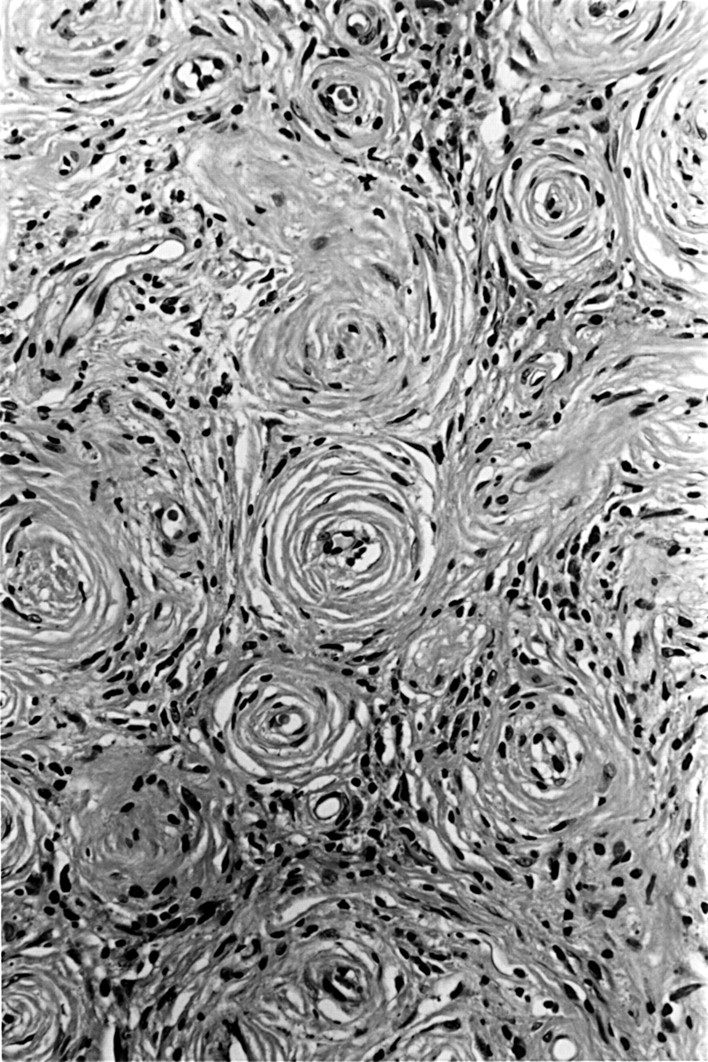
Histological examination of resected small fragments of tissue revealed the characteristic features of the fibrotic stage of EAF—perivascular whorling of bland collagen fibres, with resulting obliteration of vessel lumens (fig 1). Scattered eosinophils were present within the fibrosis. Separate fragments of normal mucosa, covered by respiratory-type epithelium, contained a moderate chronic inflammatory cell infiltrate, including numerous eosinophils. There was no evidence of vasculitis, giant cells, necrosis, or granuloma formation.

Established lesion of eosinophilic angiocentric fibrosis: perivascular fibrosis with obliteration of vessel lumens. Adjacent inflammatory infiltrate comprises eosinophils, lymphocytes, and plasma cells (haematoxylin and eosin).
Review of the previous nasal biopsy showed fragments of mucosa and granulation tissue, which contained a mixed inflammatory cell infiltrate comprising mainly plasma cells, but also numerous neutrophils, eosinophils, and lymphocytes. Focally, several clusters of foreign body-type giant cells were present (fig 2), although true granuloma formation or necrosis was not seen. Polarisable material was not identified, and stains for fungi, acid fast bacilli, and bacteria were negative. Occasional eosinophils and neutrophils traversed vessel walls, but there was no evidence of fibrinoid necrosis or vasculitis.

{kind=link}
{kind=link}
The initial nasal biopsy demonstrating giant cells, numerous plasma cells, neutrophils, and occasional eosinophils (haematoxylin and eosin).
Discussion
EAF was first described in 1985 as a specific inflammatory lesion of the nose and upper airways characterised by a predominance of eosinophils, which progresses to a fibrotic stage with a distinctive perivascular morphology. In total, six cases have been reported previously.1–4 Two further cases were mentioned in a letter by the original authors.5 Seven of the eight cases occurred in women. Patients ranged in age from 25 to 59 years. Patients present with nasal obstruction and postnasal drip, or breathlessness. The aetiology of EAF is unknown. It has occurred in association with granuloma faciale and atopy, but in most cases, the fibrotic lesion has developed after local surgery. The original report specifically rules out WG, based on the histological findings in their cases. However, only three of the previously reported cases mention the absence of circulating autoantibodies. Most cases have had a benign but recurrent course.
The inflammatory infiltrate in our original biopsy differed from previous reports. Numerically, plasma cells were the most common inflammatory cell, and were found focally in large aggregates. Increased numbers of eosinophils, some forming small clusters, were also seen. The scattered foreign body-type giant cells were also unique to our case. These findings first suggested WG, even in the absence of necrobiotic vasculitis. This diagnosis was confirmed by further clinical (flitting arthralgia and haematuria), biochemical (C-ANCA, anti-PR-3 antibodies, and raised creatinine) and radiological findings (extensive involvement of the nasal passages).
Our case is the second report of EAF occurring in a man. There is no obvious intrinsic reason why it should be more common in women—indeed, both WG and granuloma faciale are more common in men—yet reported cases thus far indicate a female preponderance. The rarity of the lesion itself might account for this seeming imbalance.
This association with WG has implications for the aetiology and pathogenesis of EAF. WG may present in a limited form, restricted to the nose and upper airways6—the same anatomical distribution as EAF. Recently, Gindre et al have shown that in the established stenotic vascular lesion of WG, type 1 collagen is laid down around vessels in concentric layers.7 Therefore, we suggest that, at least in our present case, the EAF lesion may arise as an exaggeration of this fibrotic response in WG.
Our case also challenges the existence of EAF as a distinct entity. Its increasing number of disease associations (atopy, granuloma faciale, and WG) and its progression, spontaneously or after surgery, suggests that rather than being a single entity EAF might represent an unusual reaction pattern, which can arise in several different circumstances. Because our patient also has a history of atopy (asthma) and of previous nasal surgery, it is not possible to determine the contribution of each, if any, to its development in this case.
In conclusion, we present the first case of EAF associated with WG. We suggest that instead of being a distinct disease entity, EAF might be an unusual exaggerated fibrotic reaction of the upper airways, which may arise in several situations. However, further case reports of this rare lesion are necessary before its aetiology and pathogenesis can be defined.