

Abstract
Inflammatory myofibroblastic tumor of the trachea is a rare benign tumor in adults. It is mostly seen before the age of 16. We describe a 20-year-old female patient who presented with stridor. She had a fixed obstruction on spirometry, and computed tomography and bronchoscopy confirmed tracheal thickening and stenosis below the vocal cords and bronchial wall thickening at the level of the carina. Bronchoscopic biopsy confirmed an inflammatory myofibroblastic tumor. She recovered after mechanical dilatation and resection via rigid bronchoscopy, followed by corticosteroid therapy.
Introduction
Inflammatory myofibroblastic tumor is rare, with a frequency of 0.04–0.07% of all respiratory tract tumors. It generally presents in patients less than 16 years of age.1–3 The lesion has been referred to as plasma cell granuloma, inflammatory myofibroblastic tumor, inflammatory myofibrohistiocytic proliferation, fibroxanthoma, histiocytoma, fibrous histiocytoma, xanthomatous pseudotumor, post-inflammatory pseudotumor, mast cell tumor, and plasma cell-histiocytoma.4,5 Inflammatory myofibroblastic tumors are frequently found in the lung, but similar lesions have also been reported at almost every site in the body.3 We report a 20-year-old female patient with inflammatory myofibroblastic tumor of the trachea.
Case Report
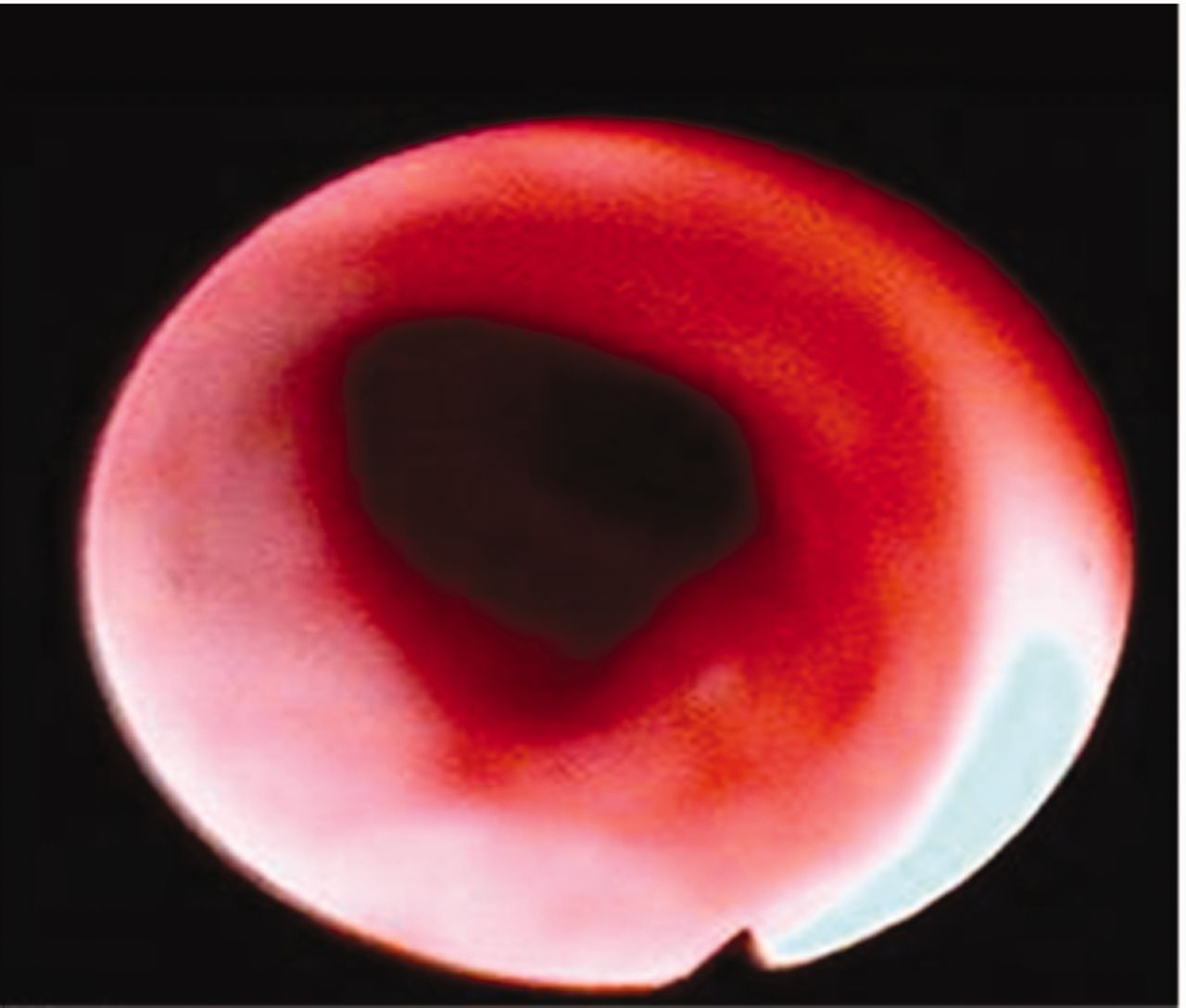
A 20-year-old female patient with unremarkable medical history was admitted to our hospital with the symptoms of hoarseness, cough (without sputum), exertional dyspnea for 1 year, and a recent wheezing attack in addition to dyspnea and cough. She was unresponsive to asthma medications, which were started before admission to our unit. Physical examination was remarkable for inspiratory stridor. Chest x-rays were normal. A pulmonary function test revealed a fixed obstruction with FEV1/FVC of 0.46, FEV1 of 62% of predicted (2.10 L), and FVC of 119% of predicted (4.60 L) (Fig. 1A). Bronchodilator reversibility of FEV1 was negative. Computed tomography of the neck and thorax revealed tracheal thickening and stenosis below the vocal cords and bronchial wall thickening at the level of the carina (Fig. 2). Diagnostic flexible bronchoscopy showed tracheal stenosis approximately 2 cm below the vocal cords, with an irregular mucosal appearance (Fig. 3).

A. Flow-volume curves (A) before and (B) after treatment.

Left: Tracheal thickening and stenosis below the vocal cords. Right: Bronchial wall thickening and stenosis at the level of the carina.

Bronchoscopy shows tracheal stenosis and irregular erythematous mucosa approximately 2 cm below the vocal cords.
Mechanical dilatation and resection, via rigid bronchoscopy, was performed in both the trachea and bronchial system. Histology reported an inflammatory myofibroblastic tumor, with S100 negativity, desmin positivity for smooth muscle cells, and trichrom positivity for collagen tissue (Fig. 4). After dilatation and resection she was started on deflazacort 30 mg/d. Two weeks later her symptoms had decreased, and at 6-month follow-up she was asymptomatic, with a normal pulmonary function test and normal thorax computed tomography. The pulmonary function test showed FEV1/FVC of 0.79, FEV of 119% of predicted (4.02 L), and FVC of 132% of predicted (5.09 L) (see Fig. 1B)

A: The polypoid tumor consisted of a proliferation of myofibroblastic cells with collagenous stroma cells with inflammatory infiltrate (hematoxylin and eosin stain, × 10). B: Widespread cytoplasmic positivity in tumor cells (vimentin stain, × 20).
Discussion
Inflammatory myofibroblastic tumor has various names, including inflammatory pseudotumor, histiocytoma, fibrous histiocytoma, xanthoma, xanthofibroma, xantogranuloma, and plasma cell granuloma.6–8 Barker et al, who reported the first case of tracheal plasma cell granuloma in the literature, used a conservative approach.9 Inflammatory myofibroblastic tumors account for 0.04–0.7% of all lung neoplasms.1,2 However, it is the most common benign lung tumor in children under 16 years of age,2,3 and is usually seen in patients less than 16 years of age.10 Inflammatory myofibroblastic tumors have rarely been reported in patients older than 16.11 Tracheal inflammatory myofibroblastic tumor has rarely been reported in adults.
A large series by Bahadori and Liebow reviewed 40 patients with inflammatory myofibroblastic tumor of the thorax. Among these patients, 15 (38%) were 1–16 years of age (mean age 8 years) and only one child had a tracheal tumor.12 To our knowledge, only 11 adult tracheal inflammatory myofibroblastic tumors have been previously reported (Table).11,13–22 Apart from patients described in the Table, Lee et al reported 15 patients (mean age 31.3 y, range 7 months to 61 y) who underwent surgery for inflammatory myofibroblastic tumors, 4 of which were tracheal,4 but the ages of those 4 patients were not stated.
Reported Tracheal Inflammatory Myofibroblastic Tumors in Adults
Because of the inconsistency in the pathology diagnosis of the tumor and the limited number of patients typically seen with inflammatory myofibroblastic tumors, the treatment of choice remains controversial. Inflammatory myofibroblastic tumors can be located in the peritoneum, liver, spleen, breast, spinal cord, brain, or respiratory system.23–28 They are more frequently seen in the lower lobe of the right lung, and form a solitary, oval, and well defined lobulated mass that is peripherally located.29–31 A mass lesion is located peripherally in 87% and centrally in 6%, and the lesions can present radiologically as multiple nodular (5%), pleural based, cavitary lesions (5%), or can present with lobar atelectasis (8%) or hilar lymphadenopathy (5%).32,33 The World Health Organization's definition is a lesion composed of a myofibroblastic spindle cell population accompanied by an inflammatory infiltrate of plasma cells, lymphocytes, and eosinophils.34 Myofibroblastic spindle cell populations account for the 70% of the total cell population in inflammatory myofibroblastic tumors, as in our patient. The symptoms are usually nonspecific, and include dyspnea, stridor, chronic cough, hemoptysis, and pleuritic chest pain. Some cases have been misdiagnosed and treated as asthma. In the small number of patients having endobronchial lesions, the clinical presentation may be acute and serious due to post-obstruction pneumonia or symptoms associated with airway obstruction.11,34 Superior vena cava syndrome was reported as a serious associated condition in one case report of inflammatory myofibroblastic tumor of the mediastinum.35
The etiology of inflammatory myofibroblastic tumor is unclear, but trauma to the affected region secondary to inflammation has been proposed. Currently, due to the existence of rare cases with a more aggressive clinical picture, including local recurrence, malignant transformation, or metastasis, it was believed that inflammatory myofibroblastic tumor was a low-grade mesenchymal malignancy.34 The findings about its recurrent chromosomal translocations involving 2p23, the anaplastic lymphoma kinase gene site, and the presence of other associated positive genetic fusions also led to the proposal that inflammatory myofibroblastic tumor was a malignant process rather than a reactive lesion.34,36–40 Applebaum et al proposed that COX2 and VEGF, as mediators of angiogenesis, might play a role in the pathogenesis and growth of inflammatory myofibroblastic tumors.41
Surgical resection has been the most relevant treatment for inflammatory myofibroblastic tumor with tracheal involvement. Surgical resection can be either intraluminal bronchoscopic removal with biopsy forceps, CO2 laser, or open surgical intervention with segmental tracheal resection. A radical surgical approach and/or adjuvant radiotherapy and chemotherapy are not indicated unless the disease is aggressive.34 Corticosteroids and non-steroidal agents have been reported as an alternative in rare selected cases.42 We performed intraluminal bronchoscopic removal and dilatation via rigid bronchoscopy, then corticosteroids.
The prognosis is usually good, but rarely this tumor may involve a local invasion. Extrapulmonary inflammatory myofibroblastic tumors have a recurrence rate of 25%. Recurrence is related to the tumor's location, resectability, and multinodularity. The metastatic rate has been reported as < 5%, and metastasis is most often seen in children with intra-abdominal tumors.34,43 Fabre et al followed 25 patients who had complete resection, with a median follow-up of 80 months (range 4–369 months), and both the 5-year and 10-year disease-free survival rate was 89%.14 The recurrence rate for tracheal inflammatory myofibroblastic tumor is unclear due to the limited number of cases.
In conclusion, inflammatory myofibroblastic tumors, particularly those with tracheal involvement, are rare in adults, and may present clinically like asthma or foreign body obstruction. Endobronchial involvement may show an acute presentation of respiratory distress. The most relevant therapy is open surgical or bronchoscopic resection. Follow-up is recommended to monitor for recurrence.
Acknowledgments
We thank Prof RW Guillery FRS, Oxford University, Oxford, United Kingdom, for editing assistance.
Footnotes
- Correspondence: Yasin Abul MD, Department of Pulmonary Medicine, Farabi Hospital of Chest Diseases, Faculty of Medicine, Karadeniz Technical University, Trabzon, Turkey 61080. E-mail: abulyasin{at}yahoo.com.
The authors have disclosed no conflicts of interest.
- Copyright © 2013 by Daedalus Enterprises





{kind=link}
{kind=link}
{kind=link}
{kind=link}