

Abstract
Relatively little attention has been directed toward damage inflicted upon the airway network that connects the alveoli, or toward the problems caused by invasive ventilation for patients with severe airflow obstruction. Mechanical ventilation with positive pressure can cause non-edematous barotrauma, inflict airway injury, and promote lung remodeling. Interactions between patient and ventilator, largely mediated through dynamic hyperinflation, include functional consequences for hemodynamics, respiratory muscle function, breathing work load, and patient-ventilator synchrony. Awareness of such associations not only helps to avoid complications during and after the critical phase of obstructive illness, but also opens a window to improved patient comfort and safety. The purpose of this review is to survey the range of structural damages and functional impairments that occur in an “obstructive” context.
- obstruction
- airway injury
- dynamic hyperinflation
- barotrauma
- mechanical ventilation
- injury propagation
- ventilator-induced lung injury
Introduction
In recent years, most discussions of ventilator-associated problems have focused on the potential for tidal volume (VT) and pressure to damage the alveoli of patients with preexisting lung injury, in a process known as ventilator-induced lung injury (VILI). Controversy continues regarding the factors that assume most importance, but it is generally agreed that high inflation pressures are dangerous, especially in the setting of heterogeneously distributed lung pathology, widespread alveolar collapse, and tidal opening and closure.1–3 By comparison, relatively little attention has been directed toward damage inflicted upon the airway network that connects the alveoli, or toward the problems caused by invasive ventilation for patients with severe airflow obstruction. The purpose of this review is to survey the range of structural damages and functional impairments that occur in an “obstructive” context.
The Airways as a Conduit for VILI Propagation
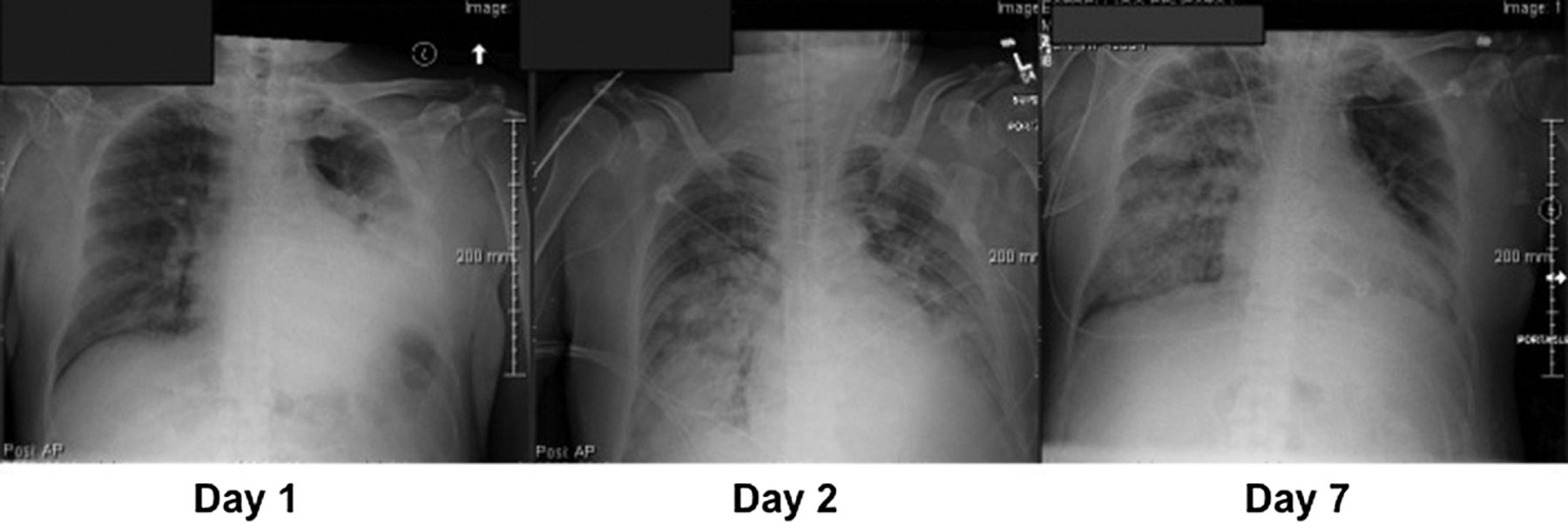
The diffuse injury of ARDS is often considered a process that begins synchronously throughout the lung, mediated by inhaled or blood-borne noxious agents. But inflammatory lung injury may also begin focally and propagate sequentially via the airway network, proceeding mouthward from distal to proximal. Such propagation could explain why lobar pneumonia often “blossoms” into generalized ARDS after intubation, even in patients with normal hearts (Fig. 1). If so, modifications of ventilatory pattern and position aimed at geographic containment of the injury process could help prevent its generalization and limit disease severity. Whereas small VT and PEEP tend to limit proximal expulsion of bio-fluids originating in the periphery, large VT and forceful exhalation efforts increase peak expiratory flows, often creating an expiratory flow bias that encourages their mouthward migration. Experimental work suggests that large tidal breaths, powered by enhanced expiratory recoil of higher volumes, are a form of ineffective coughing, without expelling secretions from the airway.4 Conversely, the rapid inspiratory peak flows typically selected for flow-controlled, volume-cycled ventilation, and inherent to the decelerating flow waveforms that accompany pressure control, encourage fluid migration in the opposite direction.5,6 Patterns of high-frequency ventilation characterized by long inspiratory/expiratory ratios that produce an expiratory flow bias have been associated with mucus transfer toward the airway opening. Efficacy of percussive ventilation in clearing secretions relies on an asymmetric flow profile that favors expiration.7

Propagation of pneumonia and lung injury. On day 1, pneumonia originating in the left lower lobe was addressed by intubation, mechanical ventilation, antibiotics, fluid repletion, and routine side-to-side variation of positioning. Despite appropriate treatment, by day 2, the process had become primary (pulmonary) ARDS. By day 7 the original pneumonia site had cleared, whereas the right lung was still involved by the acquired inflammation.
Using conventional frequencies in a 2 compartment test lung system, Volpe et al demonstrated parallel findings.8 Differing compliances encouraged transfer of a mucus simulant from one ventilated test lung compartment to another across their shared carinal divider. The potential for adverse ventilatory patterns to rapidly transfer thin proteinaceous fluids mouthward in vivo was shown in a well designed experimental study9 in which radio-labeled albumin dispersed quickly and evenly throughout the lung containing the injected lobe, as well as propagated into the opposite lung soon after large VT was initiated from low PEEP. In contrast, the tracer remained within the injected lung without detectable contralateral spread when higher PEEP was used with smaller tidal excursions.9
Appropriate gravitational repositioning forms a central tenet of therapeutic postural drainage and chest physiotherapy. Conversely, to avoid unwanted drainage, precautions are taken to prevent large liquid-filled cavities (eg, abscesses) from discharging their contents into unaffected zones. Similar principles of fluid mobilization by gravity may apply on a smaller scale. If so, dependent positioning of initially focal damage could mitigate injury propagation early in the disease course, when lung liquids are thin and highly mobile.10 In the periphery, gas velocities during routine tidal breathing are very slow, and with cough and mucociliary action impeded, gravitational advantage is needed to bring secretions more centrally, where faster flows have the potential to move them. In an important recent contribution, Schortgen and colleagues11 examined the impact of position and ventilatory pattern on the dissemination of bacteria from the right lungs of rats inoculated with bacteria. Two hours of mechanical ventilation were sufficient to seed the liver, spleen, and opposite lung, but to differing extent, depending on the selections for PEEP and position. Low PEEP, high VT, and non-dependent positioning of the infected right lung promoted left lung contamination, when compared with spontaneous breathing.
Physiologic reasoning, coupled to experimental evidence, supports the potentially important place of transairway transfer of mobile, noxious biofluids. Awareness of the ventilatory and positional modifications of practice that help in containment may help avert dissemination of initially focal disease.12
Non-Edematous VILI
During the first 3 decades of using positive-pressure ventilation for life support, the varied forms of radiographically evident barotrauma were encountered frequently, and in diverse patient populations.13–15 Serious consequences ensued, and the incidence of death related to ventilator-caused air leaks was clearly unacceptable. As understanding evolved, the underlying cause appeared linked to high inflation pressures resulting from the caregiver's insistence on maintaining normal values for CO2 and pH. It is a sobering fact that in ventilated patients with asthma, for example, the rates of pneumothorax and mortality plummeted once permissive hypercapnia was adopted,16 a full 5 years before a similar strategy was promoted for ARDS.17
In the often heated discussion regarding the most important contributing mechanical cause of VILI—elevated plateau pressure or insufficient PEEP to keep the lung near fully recruited—the patient with obstructive disease may have offered an overlooked clue to resolving the issue. High plateau pressures are frequently encountered in decompensated asthma and COPD, but overt lung edema seldom results, presumably because elevated total PEEP is the primary cause of plateau pressure elevation, and driving pressure in such patients (the difference between plateau and total PEEP) usually remains modest. Therefore, while disease pathologies and mechanics are clearly different, it would seem that current ventilation approaches for acute airflow obstruction and lung-protective ARDS result in patterns that end up being remarkably similar. Both involve constrained plateau pressures and higher total PEEP (one PEEP unintentional and the other deliberate). Both strategies may avoid parenchymal edema and inflammation. This is not necessarily so with other forms of structural damage to the lung, known collectively as barotrauma.13
Airway Injury
Mechanisms of Barotrauma.
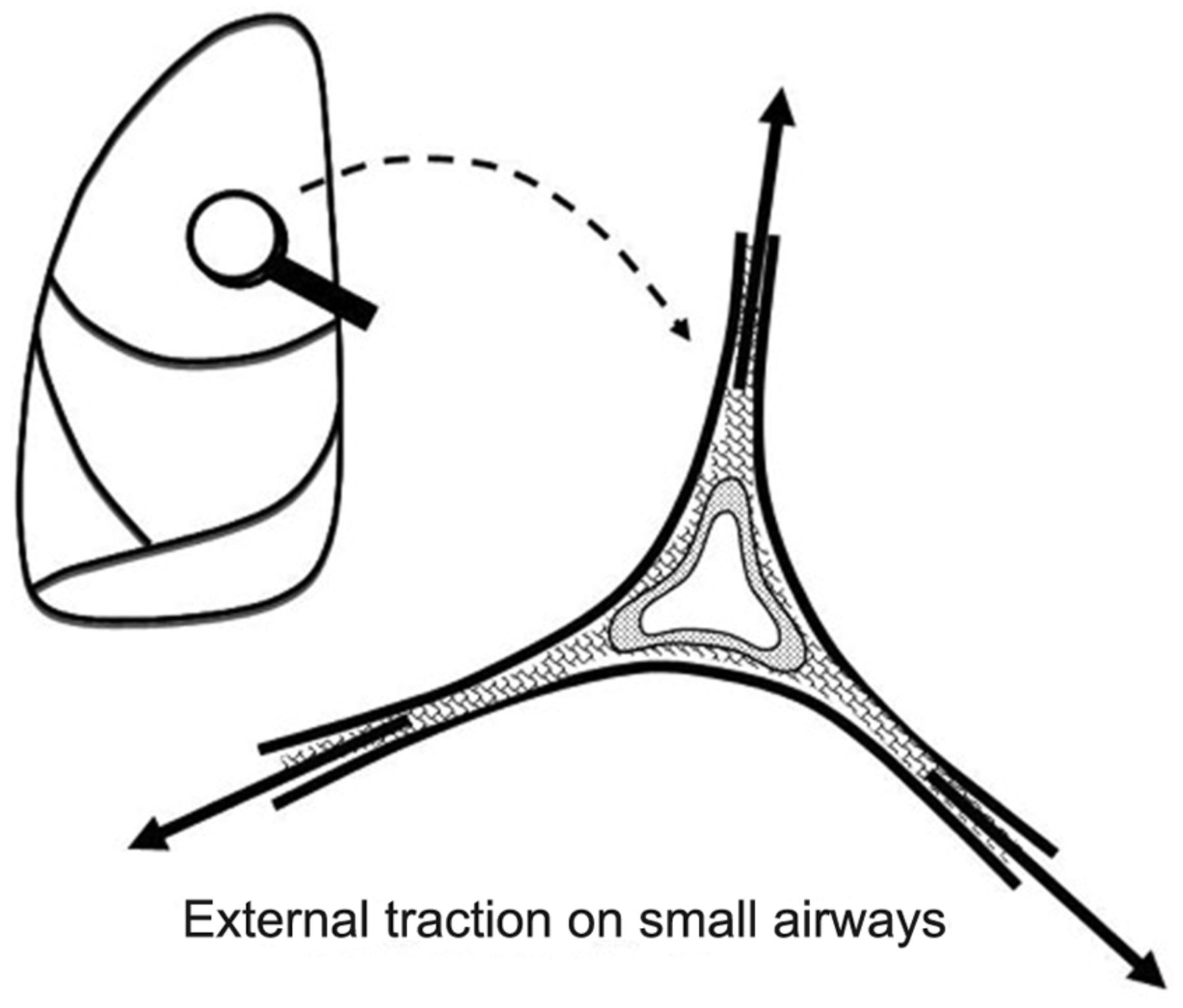
Newer data generated by quantitative computed tomography suggest that alveolar overstretching tends to manifest when the VT and PEEP more than double the aerated functional residual capacity.18,19 The resulting overstretch ruptures the matrix and may threaten to disrupt the fibrous skeleton of the lung. Rupture is especially likely when there is force amplification (stress focusing) within a heterogeneous lung20,21 or weakened integrity of the interconnecting tissues. Intolerable forces may cause regional end-inspiratory strains that rupture the fragile jeopardized alveoli (Fig. 2). Although the peak airway cycling pressure has been cited frequently as the most important risk factor for ventilator-related barotrauma, clearly it is not the only one22–27 (Table 1).
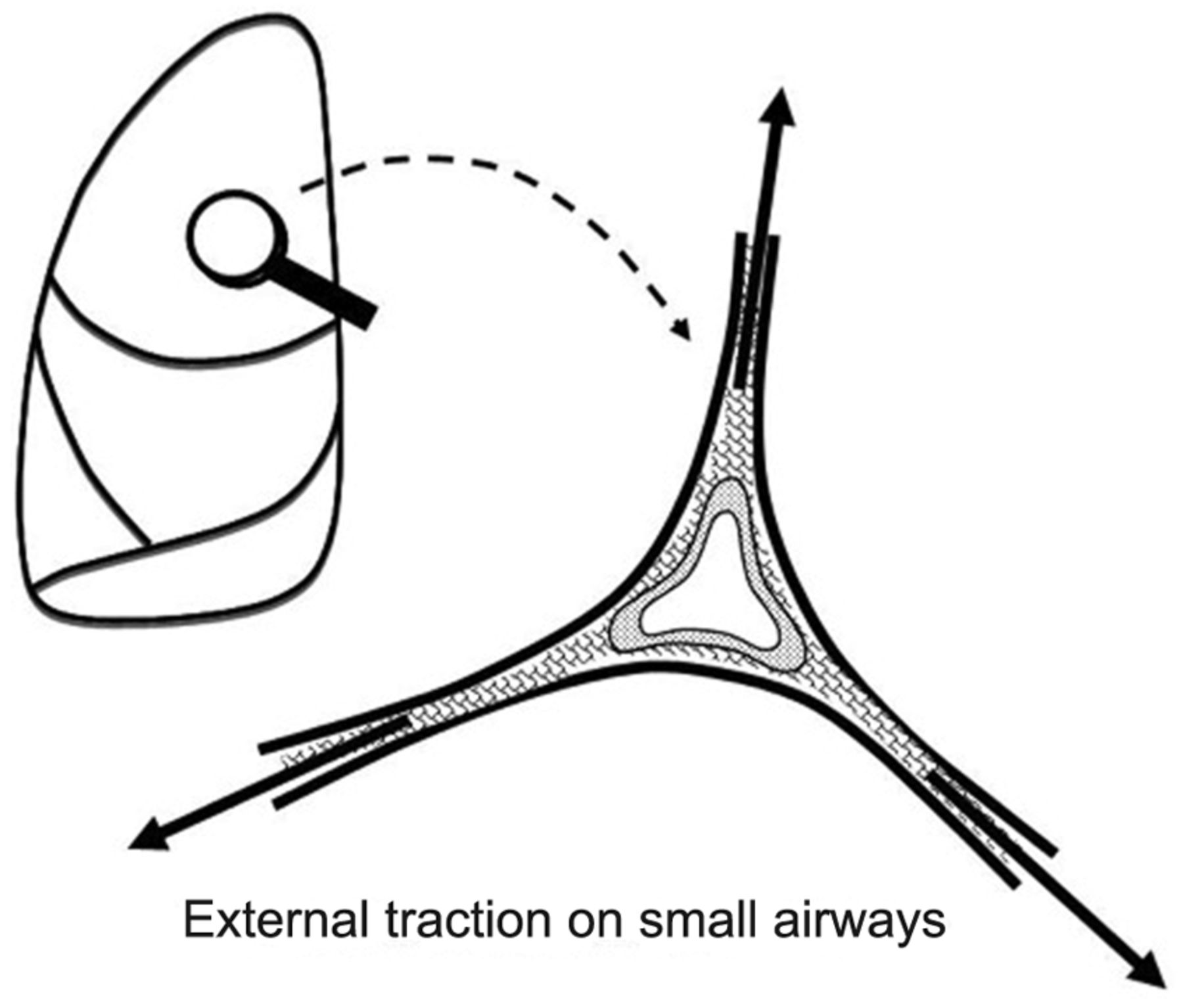
Amplified tissue tension at the junction of the relatively fixed small airway and surrounding expanding alveoli may apply damaging force to delicate structures.
Predispositions to Barotrauma
The peak dynamic and static (plateau) airway pressures seem to contribute most to the multivariate risk. Peak dynamic airway pressure can be reduced by improving lung compliance, reducing VT or PEEP, lowering airflow resistance, or slowing peak inspiratory flow. The correlation between peak airway pressures or PEEP and barotrauma is not a tight one.22,23 A necrotizing parenchymal process, inhomogeneous lung pathology, young age, excessive airway secretions, and duration of mechanical ventilation are major predispositions.23,25 Surface blebs can give way suddenly, whereas parenchymal rupture seems to require sustained hyperexpansion of fragile alveoli. Therefore, the mean alveolar pressure, averaged over an entire respiratory cycle, may be an important contributing factor. As major determinants of peak and mean alveolar pressures, minute ventilation requirement and high levels of PEEP contribute to the hazard.22,24 PEEP itself may contribute little to the risk of barotrauma if it is applied within the range over which lung recruitment is its primary action. Further, PEEP may also help to even the distribution of airway pressures and to reduce stress focusing that occurs when units with heterogeneous mechanical properties are juxtaposed.
Mechanical heterogeneity plays a central role in the stress and strain of mechanical ventilation. Even though the ventilated diseases of airflow obstruction—exacerbated COPD and acutely decompensated asthma—are often viewed as homogeneous and diffuse, they are not. Here mechanical heterogeneity is caused less by variations of compressive lung weight, blood flow distribution, and pleural pressure difference (as in ARDS) and more by heterogeneity of time constants due to regional differences in airway secretions, airway inflammation, bronchoconstriction, chest wall characteristics, and gas trapping.28–30
On first consideration it might seem that end-inspiratory static pressure (the pressure that acts in conjunction with thoracic compliance to determine overall lung volume and average alveolar stretch) should correlate even more closely with pneumothorax than with peak dynamic ventilator cycling pressure (peak dynamic pressure). Plateau pressure does bear a somewhat stronger relationship to pneumothorax than does peak dynamic pressure. However, airway resistance varies greatly among the bronchial channels of a non-homogeneously affected lung, so that increasing the dynamic pressure within the central airway may encourage regional-over distention and alveolar rupture in channels with open pathways to weakened alveoli. Moreover, rapid inspiratory flow tends to drive secretions away from the airway opening8 and even injure airway epithelium.31 It stands to reason that raising the peak flow to lengthen expiratory time is not risk-free. Improving airway resistance or lung compliance and reducing VT and PEEP are preferable methods for lowering peak dynamic pressure.
There does not seem to be a sharp threshold value of peak ventilator cycling pressure below which alveolar rupture fails to occur.22,23 As a rule, however, pneumothorax becomes much more likely at peak ventilator cycling pressures > 40 cm H2O.22 A peak tidal pressure > 35 cm H2O usually achieves or exceeds the alveolar volume corresponding to total lung capacity in a patient with a normal chest wall. Conversely, when the chest wall is stiff, high plateau pressures may be better tolerated. Secretion accumulation, blood clots, or foreign material can increase the degree of mechanical non-homogeneity or create ball-valve phenomena that increase the barotrauma hazard (Table 2)
Preventing Ventilator-Related Alveolar Rupture
Pathogenesis of Small Airway Damage and Alveolar Rupture.
Radiographically evident barotrauma—interstitial emphysema, pneumomediastinum, pneumoperitoneum, subcutaneous emphysema, cyst formation, and pneumothorax (pneumothorax)—are among the highly feared iatrogenic consequences of ventilatory support.24 Direct rupture of the visceral pleura undoubtedly can occur as a consequence of distention by positive pressure, but barotrauma that complicates mechanical ventilation can also develop by a more circuitous path. Rupture of weakened alveolar tissues is particularly likely to affect “nonpartitional” or “marginal” alveoli, which have bases contiguous to relatively immobile structures—vessels, bronchioles, or fibrous septae.32,33 Alveolar pressures rise disproportionately more than interstitial pressures during ventilation with high pressure, during severe coughing with plugged small airways (ball valving), or during blunt chest injury that occurs with the glottis closed, producing non-physiologic pressure differences between marginal alveoli and the contiguous perivascular connective tissues. Once rupture occurs, extra-alveolar gas follows a pressure gradient down the path of least resistance, migrating along the perivascular sheaths toward the hilum. The gas continues to track centrally, forming a pneumomediastinum that may or may not be evident on routine films.
In the absence of preexisting mediastinal pathology, extra-alveolar gas dissects through loose fascial planes, usually decompressing into the soft tissues of the neck (subcutaneous emphysema) or, more rarely, retroperitoneum (pneumoperitoneum). Pneumothorax occurs in a minority of such cases (perhaps 20–30%) when soft-tissue gas enters the pleural space via an interrupted or weakened mediastinal pleural membrane. Interstitial emphysema, pneumomediastinum, and subcutaneous emphysema typically have little hemodynamic importance and seldom substantially affect gas exchange in adults without airflow obstruction. However, life-threatening airway compression may ensue if extravasated gas cannot fully decompress into the soft tissue sink.34–36 Because their presence signals alveolar rupture and the potential for pneumothorax to evolve, these signs are important to detect in the ventilated patient. The consequences of pneumothorax for the ventilated patient are severe, well known, and will not be reviewed here.
When normal bronchovascular channels that lead to the mediastinum are blocked by structural distortions related to disease, interstitial gas accumulates locally or migrates distally to produce subpleural air cysts that compress parenchymal vessels, create dead space, increase the ventilatory requirement, and cause major problems for ventilation-perfusion matching.34,35 Although presumably important, the exact roles of secretion plugging and “ball valving” are not well defined in this process of violent and poorly modulated parenchymal remodeling. What is clear, however, is that sudden development of cysts is an ominous finding that usually presages tension pneumothorax a short time afterward.35
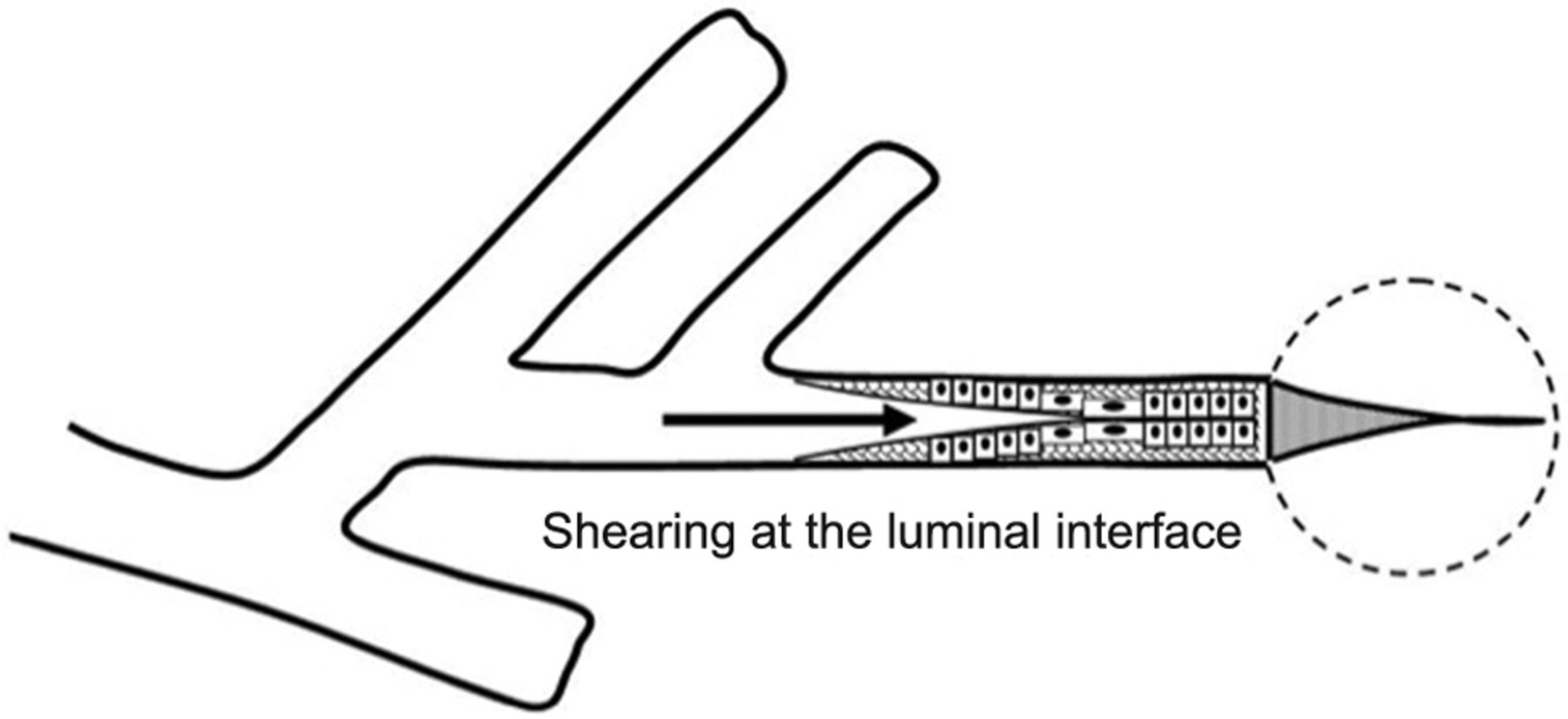
Direct Bronchopulmonary Injury. Until quite recently, the development of ventilation-caused endobronchial damage was believed to occur rarely in adult patients. Autopsy studies of patients ventilated at moderately high pressures for extended periods, however, have demonstrated that small airways unsupported by cartilage can sustain considerable damage at high airway pressures,37 especially when central airway pressures are felt in full force by lung units that remain unopened (Fig. 3). Such a mechanism is believed to contribute to neonatal bronchopulmonary dysplasia when insufficient PEEP is used in the face of high airway pressure, and perhaps to the failure of high-frequency ventilation to protect against it.38,39 Repeated application of high shearing forces developed along the epithelial surfaces of the terminal bronchioles in the process of opening and closure is believed to incite inflammation31 (Fig. 4).
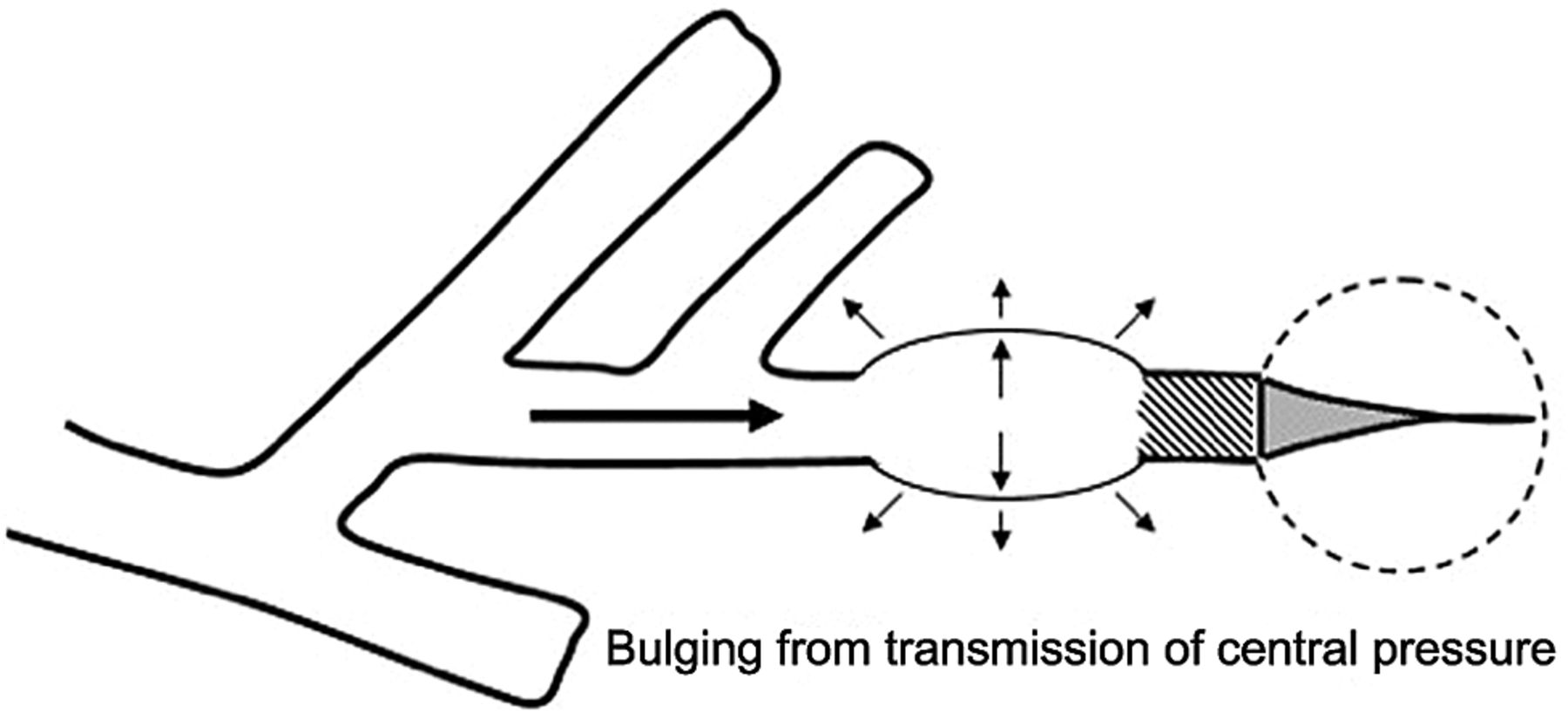
Bulging of unsupported small or terminal airway “dead-ended” by nonexpanding parenchyma.

Epithelial shearing forces repeatedly distort the lining cells of terminal airways during tidal inflations and may thereby signal inflammation.
Micro-Cystic Barotrauma. Airway distortion predisposes to cystic parenchymal damage, disordered gas exchange, and impaired secretion clearance. Microcysts occur throughout the lungs of patients with ARDS ventilated with high inflation pressures, and these tend to prevail in the dependent zones where high risk “junctional” interfaces between aerated and non-aerated units are most prevalent.40 Yet, larger, coalescent, and emphysema-like distortions have been shown to develop in open non-dependent regions exposed continuously to high “remodeling” (structure altering) forces applied to an inflamed lung.41–44
Macroscopic cystic barotrauma, once common, is fortunately a rare occurrence now, due to modern day use of lower airway pressures. But it remains a risk in young patients with necrotizing pneumonitis, narrowed airways, and retained secretions who are ventilated imprudently.35,43,45 Alveolar rupture and focal gas trapping are key to its pathogenesis. As predicted by the Law of Laplace (P = 2T/R), the pressure (P) required to maintain a fixed tension (T) in the wall of a spherical structure falls as its radius (R) increases. Therefore, once started, the process is self-reinforcing, so that a cyst created by positive airway pressure may grow to large dimensions over only a few days. As parenchymal cysts of this type evolve, they compress normal lung tissue, stiffening the lung and further increasing the airway pressure needed for effective ventilation. Furthermore, blood flow diverts away from areas of cyst expansion, creating dead space that increases the ventilatory requirement and therefore the mean alveolar pressure. Effective secretion management, treatment of infection, and, most importantly, reduction of airway pressure are fundamental to effective management.
Systemic Gas Embolism. For patients with ARDS ventilated with high tidal pressures and maintained with relatively low left ventricular filling pressures (“wedge pressures”), peak and mean alveolar pressures may exceed pulmonary venous pressures in certain lung regions. If alveolar rupture opens a communication pathway into the vascular system, this pressure difference from air space to pulmonary vein may on rare occasion drive air into systemic circulation. Micro-bubbles can then cause vasospasm, neurologic irritation, seizures, and damage, or myocardial infarction.45,46 Usually, the infarction is inferior, as the buoyant air percolates into the right coronary artery, which lies anteriorly and superiorly in the supine position. Regionally constrained livedo reticularis (mottled reticulated vascular pattern) is a rare signature skin finding.45
Functional Impairment in Ventilated Patients With Obstructive Disease
Numerous adverse interactions may develop between the ventilator and patient with airflow obstruction. The root causes of these adverse events relate primarily to the necessity for airway intubation in a patient dependent on effective airway hygiene, and to the dynamic hyperinflation that occurs during ventilator support. As it does for everyone, intubation interferes with the native efficiency of coughing and secretion clearance. The potential consequences for patients who produce copious airway secretions are especially severe. Adequate hydration, bronchodilation, inflammation suppression with short course corticosteroids, as well as prevention and treatment of infection are of clear importance in this setting and will be mentioned only in passing. It is the consequences and management of dynamic hyperinflation that more often evade detection or are inexpertly managed.
Dynamic Hyperinflation (Air Trapping)
Expiration is normally a passive process that uses elastic energy stored during inflation to drive expiratory airflow. If the energy potential stored during inflation is insufficient to return the system to a relaxed equilibrium before the next inspiration begins, flow continues throughout expiration and alveolar pressure remains positive at end-expiration, exceeding the clinician-selected PEEP value.47–51 This unintended supplemental positive distending pressure within the alveoli at end expiration (auto-PEEP also known as intrinsic PEEP [PEEPi]) both increases the driving pressure for expiratory airflow and augments recoil, thereby helping to overcome airflow resistance. The need to hyperinflate is generated by a long expiratory time constant (mathematically, the product of expiratory resistance and compliance), increased ventilation requirements, and inadequate exhalation time.
Though essential for exhaling the VT, hyperinflation has adverse physiologic consequences as well. Because the hyperinflation-related end-expiratory alveolar pressure encourages deflation, it must first be counterbalanced before inspiration can begin.52–55 Thus PEEPi adds to the other components of the equation of motion to elevate the mean inflation pressure and inspiratory work of breathing.53 The process of air trapping contributes to an increase in the respiratory work of breathing in at least 2 other ways. Hyperinflation drives the respiratory system upward toward the least compliant portion of the pressure-volume relationship, incurring increased elastic work per liter of ventilation.56 At these higher volumes the lung approaches its elastic limit as the recoil tension of the distended rib cage becomes expiratory rather than inspiratory in nature.57 Hyperinflation also tends to convert more of the well perfused (“zone 3”) lung into less well perfused tissue, thereby increasing ventilatory dead space and the minute ventilation requirement.
When dynamic collapse of the airway occurs during tidal ventilation, and breathing requirements are high, there is little alternative to hyperinflation, CO2 retention, or both. At the chosen level of minute ventilation, maintaining the lower lung volume may be either too energy costly or physically impossible. For this reason, many patients with severe obstruction do not or cannot decrease their lung volumes when recumbent.58 Dyspnea is experienced by most patients with severe airflow obstruction on assuming horizontal positions.
Heterogeneity of Auto-PEEP.
Diverse mechanical environments often coexist within the diseased lung (Fig. 5). The distribution of gas trapping varies regionally, depending on the local mechanical properties of the airways. Therefore, at the end of the expiratory cycle some zones remain patent, and some have sealed much earlier in the deflation cycle (see Fig. 5). The natural tendency for dependent airway closure and gravitational pooling of airway secretions is exacerbated by recumbency. Dependent airway closure tends to occur before end-expiration, trapping gas at alveolar pressures that exceed the measurable PEEPi.52,59 During volume-controlled ventilation, plateau pressure tracks hyperinflation more reliably than direct measurements of PEEPi.60 Because of such “early” airway closure, measured auto-PEEP may dramatically underestimate dynamic hyperinflation and barotrauma risk in asthmatic patients.61

Spectrum of regional auto-PEEP values (left) and their responses to adding 8 cm H2O PEEP (right). Adding PEEP may help even the disparate end-expiratory pressures and thereby improve distribution of ventilation as well as the ease of breath initiation. (From Reference 52, with permission.)
Clues to the presence of regional early closure are often seen when the airway is occluded at end-expiration. With all airways open, an unambiguously stable value of PEEPi is achieved within 2–3 seconds of airway occlusion.49,52 In the presence of early closure, the auto-PEEP value shows an atypically slow rise to its final value as quasi-occluded small airways decompress into the common airway.52,62,63 This is not the case, however, when widespread dependent airway closure occurs, as gas trapped in these zones gradually decompresses into the central airways, raising PEEPi with a characteristic pressure wave signature that continues to slowly rise for many seconds after occlusion (Fig. 6).52,64,65 It is not surprising, therefore, that regional trapping and its slowly rising waveform signature can be eliminated by raising PEEP.

Characteristic post-occlusion airway pressure signatures of homogeneously and non-homogeneously distributed auto-PEEP. (From Reference 52, with permission.)
One implication of zonal airway closure is that sitting, supine, and lateral decubitus positions may be associated with different degrees of dyspnea, gas trapping, flow limitation, and PEEP responsiveness.52,59 The need to avoid positional volume loss and gas trapping helps to explain orthopnea experienced by severely obstructed patients, despite the absence of heart failure.
PEEP on Auto-PEEP.
The cautious application of PEEP to the circuit of a patient with airflow obstruction may reduce the work of breathing if and only if tidal breaths are flow limited.51–53 In this circumstance, PEEP less than the critical closing pressure added downstream simply replaces a nearly proportional amount of auto-PEEP without causing further hyperinflation. This relieves the important component of inspiratory pressure otherwise needed to draw the breath against the expiratory bias of auto-PEEP (triggering effort) and may improve rhythm synchrony with the ventilator.
Increased Minute Ventilation Requirement.
Ventilation-perfusion mismatching is widespread in patients with severe airflow obstruction, reducing the efficiency of carbon dioxide elimination. It is not uncommon for the resting minute ventilation requirement to exceed 12 L/min (twice the normal value) in patients with exacerbated asthma or extensive emphysema and strong chemical drives to breathe. Enormous increases in the oxygen consumption rate of the ventilatory muscles have been observed in patients with obstructive lung disease. During exacerbations, the oxygen consumed by ventilation and the metabolic demands associated with heightened vigilance, agitation, or anxiety may double the total body oxygen consumption observed during fully supported breathing.66 The prevalent combination of impaired ventilation-perfusion matching, hypoventilation, and diffusion impairment results in arterial oxygen desaturation that generally responds well to modest supplementation of inspired oxygen, except when there is extensive plugging of small airways.
Reduced Mechanical Efficiency.
In patients with exacerbated COPD or decompensated asthma, the oxygen consumed in the ventilatory task is disproportionate to the amount of mechanical work actually performed. The muscles of the hyperinflated ventilatory system are inefficiently aligned, force-length relationships of the shortened end-inspiratory fibers are suboptimal, and normally efficient coordination among the various muscles of the ventilatory group is often disrupted.52,53,57 The energy cost of breathing, therefore, is greatly increased for the pressure and mechanical work actually generated by the breathing effort. Panic reactions occur, in which the desperate patient increases minute ventilation and oxygen demand, and trapped gas results in higher work load and reduced capability. The objective of ventilation should be to establish and preserve gentle breathing efforts. Pharmacologic paralysis, though sometimes needed to reduce O2 consumption and CO2 production, enables small VT and permits tolerance of hypercapnia, but must not be sustained for longer than absolutely necessary. There is clear evidence that the process of atrophy begins earlier than 18 hours when breathing efforts are completely silenced.67
Hemodynamic Compromise.
In some patients, especially those who remain passive during ventilatory support, the problems presented by air trapping and dynamic hyperinflation are as much cardiovascular as pulmonary in nature.49 A relatively high fraction of the resulting positive alveolar pressure is transmitted to the pleural space, where it impedes venous return and confuses interpretation of hemodynamic pressure measurements of central venous pressures. Lung distention also adds to pulmonary vascular resistance, exacerbating the tendencies of patients with cor pulmonale toward low cardiac output and hypotension. Marked respiratory variation of systolic and pulse pressures during passive inflation indicates physically adverse cardiac loading and strongly implies the possibility of dynamic hyperinflation.68 Patients who breathe spontaneously lower the inspiratory pleural pressure dramatically, forestalling the drop in venous return that occurs once such efforts are silenced.69
Pressure Targeted Modes and Auto-PEEP.
Pressure-targeted modes of ventilation, exemplified by pressure control, airway pressure release ventilation, and pressure support, have become increasingly popular. Because the development of auto-PEEP reduces the pressure difference between airway opening and alveolus that drives inspiration, both auto-PEEP and the size of the VT vary with airway mechanics and with the pattern of breathing and minute ventilation. For a fixed value of applied airway pressure, inspiratory VT in patients with airflow obstruction will be more sensitive to the frequency and the inspiratory time fraction (an expression of the inspiratory/expiratory ratio) than are normal subjects or those with restrictive disease.70 If the patient is passive or the amount of inspiratory muscle force remains constant, delivered VT falls as the auto-PEEP builds. This auto-PEEP/driving pressure interaction may result in triggering inconsistency and wide variability (even chaos) in breathing rhythm.71,72 The consequences for comfort and sleep efficiency are likely to be important, but clinical data are lacking on these issues at this time. Although the interactions between gas trapping and pressure targeted modes—new and old—are both intriguing and numerous, this is a topic unto itself and beyond the focus of this review.
Is Auto-PEEP Always Pathological?
The simple answer is no. Auto-PEEP with dynamic hyperinflation will be generated when minute ventilation is high and the patient is passive, even if lung mechanics are normal or fundamentally restrictive.73 Expiratory muscle activity may prevent dynamic hyperinflation while contributing to increased end-expiratory pressure. Within the “baby lung” of ARDS, there is a disconnect between the status of the lung and chest wall during auto-PEEP. Measured functional residual capacity is almost invariably reduced (even if regional volume is excessive), while the chest wall is of normal or reduced dimension—not distended. Even when the lungs are mechanically normal, if the chest wall is unusually stiff, auto-PEEP may not result in an elevated functional residual capacity. Therefore, auto-PEEP is not always pathological or even necessarily wedded to dynamic hyperinflation. In fact, the physiologic links between auto-PEEP and applied PEEP have encouraged the exploitation of auto-PEEP in maintenance of open airways in non-obstructive diseases such as ARDS. Inverse ratio ventilation and airway pressure release ventilation attempt to maintain a baseline level of PEEPi, generated by imposing brief expiratory periods.74,75 Oxygenation may benefit from the higher mean airway pressure that results from an extended machine inflation phase, without incurring end-expiratory lung collapse. In a perfectly uniform lung, PEEP and PEEPi would be interchangeable. However, in a diseased lung, heterogeneous time constants ensure that auto-PEEP will vary from site to site, with the higher values incurred where they are least needed—the most compliant units. Because applied PEEP is uniformly distributed, adjustable, and invariant with level of ventilation and position change, it is generally to be preferred to auto-PEEP for such applications. PEEP added to auto-PEEP in a patient with tidal flow limitation will help to reduce the associated triggering effort and work of breathing, as well as help to even the distribution of ventilation throughout the heterogeneous lung.
Summary
Mechanical ventilation with positive pressure can cause non-VILI barotrauma, inflict airway injury, and promote lung remodeling. Interactions between patient and ventilator, largely mediated through dynamic hyperinflation, include functional consequences for hemodynamics, respiratory muscle function, breathing work load, and patient-ventilator synchrony. Awareness of such associations not only helps to avoid complications during and after the critical phase of obstructive illness, but also opens a window to improved patient comfort and safety.
Footnotes
Dr Marini presented a version of this paper at the 51st Respiratory Care Journal Conference, “Adult Mechanical Ventilation in Acute Care: Issues and Controversies,” held September 7 and 8, 2012, in St Petersburg, Florida.
The author has disclosed no conflicts of interest.
- Copyright © 2013 by Daedalus Enterprises
References
- 1.↵
- 2.
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.
- 27.↵
- 28.↵
- 29.
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.
- 49.↵
- 50.
- 51.↵
- 52.↵
- 53.↵
- 54.
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
Discussion
Hess:
What about the use of an expiratory flow bias, which you and some others have talked about, at least from an experimental basis? Is that helpful? Is it something we should be thinking about: trying to adjust the inspiratory flow on the ventilator so that it's lower than the peak expiratory flow bias?
Marini:
Yes. The inspiratory to expiratory peak flow ratio seems to be important in driving central secretions either toward the patient or toward the airway opening.
Hess:
I think you were a co-author with Volpe?1
Marini:
You're right, Dean. Marcia did that experimental bench study with us. We also have some data from animal lungs as well. The basic idea here is that we routinely use high peak tidal flows that favor inspiration. If you use a decelerating waveform, short inspiratory/expiratory ratio, small VT, and PEEP, on inspiration the flow is fast, and because the secretions shear in the direction of flow, the high inspiratory flow drives secretions deeper into the lungs. During exhalation, because you're using small VT, you don't have a lot of recoil pressure to generate high peak expiratory flow, so you don't push the secretions mouthward. The repetitive tidal packing that you get with decelerating waveforms and pressure-controlled waveforms may be contributing to secretion retention in some patients.
Branson:
John, why is it valuable to know the FRC [functional residual capacity] and the ΔFRC and ΔPEEP in either direction? Secondly, in light of the issues of spontaneous breathing, what's your stance on APRV [airway pressure release ventilation] in ARDS? I'm asking because you talked about spontaneous breathing and the rapid increases in volume, and I thought, well, I doubt that anybody is using APRV in COPD.
Marini:
The FRC question is important. When we ventilate a patient with ARDS, I believe we should either estimate the size of the baby lung or know the transalveolar pressure in the jeopardized areas. For that purpose, transpulmonary pressure comes close, but it doesn't do the full job. Knowing the gas volume into which you push a VT allows you to calculate what Gattinoni calls the strain ratio, which is VT plus PEEP times compliance of the lung, divided by resting FRC. If you know the FRC, you have a running shot at knowing how much lung strain you're generating. I think it would also help when assessing whether the patient is getting better or worse. If the FRC tracks in one direction over time, it can give you a helpful clue that improvement or deterioration is going on.
What is happening to the FRC is one of the missing pieces, along with esophageal pressure, of a noninvasive, full-spectrum battery of lung mechanics for the bedside. You can't measure FRC in everybody. If they're vigorously and spontaneously breathing or chaotically breathing, you're not going to get a reliable, high quality automated FRC. That's a challenge worth addressing.
Hess:
My understanding from reading and listening to Gattinoni is that, if you want to get the strain ratio you, have to measure FRC on zero PEEP. My concern has been how safe is it to take away the PEEP to measure the FRC and get the strain ratio?
Marini:
I think that, like all good scientists, he is exploring an issue and modifying his viewpoint as time goes on. Luciano is talking about the relaxed lung versus the lung with the PEEP and VT in it, so his equation isn't just VT divided by FRC, which would require zero PEEP, but VT plus PEEP times lung compliance in the numerator, which gives you the extra volume component related to PEEP over and above a relaxed baseline, divided by FRC.
MacIntyre:
But do you still have to go to zero PEEP to get the FRC?
Hess:
That's what I've heard him describe.
Marini:
Yes, if you're measuring FRC you'd have to measure it at zero PEEP or correct for the PEEP-related component. You could measure FRC on PEEP and subtract away the PEEP component. FRC in the denominator has to be at an equivalent of zero PEEP, but if you know PEEP and you have an estimate of lung compliance (which you can get with transpulmonary pressure), you can then subtract off what you need and infer what the baseline FRC would be. And now to go back to the APRV question …
Branson:
My question is about the idea that APRV is safe because the system is able to limit pressures. Based on what you've demonstrated, with the patient taking large tidal volumes during spontaneous breathing, what's your opinion about that?
Marini:
My opinion is probably like yours, Rich, that it's a nice idea, and it's conditionally correct that APRV is safe, so long as the patient isn't making violent movements or tugging with inspiratory forces that add to the high resting airway pressure that you maintain. And then you've got rapid release of volume and rapid buildup of volume. Such large and rapid changes in volume might contribute to problems.
MacIntyre:
John, I want to go back to the ARDS lung. Clarify this stress and strain concept you and Gattinoni speak about. I got the strain part with change in volume and referencing it to FRC, but the stress part I'm still confused about, because if you have a normal lung and you apply pressure of 35 cm H2O, the lung physically stretches and enlarges. But if you get a really stiff lung and you apply 35 cm H2O, it may not stretch very much or physically expand. Is the 35 cm H2O in the injured lung that's not being stretched very much as dangerous as the stress in a normal lung that is being stretched?
Marini:
I think we get lost in the single compartment, “balloon on a tube” model. You're right that a stiffer lung will not expand very much overall, but at the microscopic level you're playing a different game. There it's the transalveolar pressure, and that can be very high, especially at focal stress points. Gattinoni looked at the specific compliance of the tissue in ARDS and found that it remains relatively normal. If that is correct and your chest wall is not stiff, you're going to cause damage with high airway pressure.
MacIntyre:
So the stiff lung that's not expanding is as much at risk as a normal lung that is expanding?
Marini:
Do you mean a stiff lung unit, or the global lung?
MacIntyre:
The global lung.
Marini:
Yes.
MacIntyre:
I thought I understood it, but I wanted to hear it from the expert.
Kallet:
John, you had mentioned the spreading of infection by positioning, and rapid pressure changes driving fluids deeper into the lung. We use a lot of prone positioning in patients with ARDS, some of it thanks to you. But the people who make the beds have their own protocol they're always trying to sneak in. Often I'll come in to see somebody who's barely oxygenating, and they have these radical position changes back and forth. It bothers me just in terms of trying to stabilize the patient's oxygenation and figure out what's going on. Do we know if maybe in early ARDS or early pneumonia, with thin secretions, that this might not be a good idea? How should we deal with that?
Marini:
I wish I had the experimental backup to answer that question. We do know that we can propagate secretions through the lung just by a simple position change and ventilation.2 We think the concept is valid. We also know we can protect against migration with a little bit of PEEP and a little bit of inclination: not radical positioning.
What role this plays I can only tell you anecdotally. I've had patients I have intentionally kept at 15 or 30 degrees, injured side down and head slightly up, with PEEP of 5 to 10 cm H2O. That's all you really need to inhibit the secretions, unless there are copious secretions due to volume overload or the patient's condition. That's how you can protect against propagation for the 12 to 24 hours it takes until the gels form and secretions become less dangerous as a propagation threat. We do not have data on what to do. With someone with highly lateralized disease I would not turn them good lung down; that's been reported in the literature, and I think it could be dangerous, or at least counterproductive.
Kallet:
In California, bed sores are a reportable incident to the state; it's a very big deal, and nursing is understandably worried about not performing routine position changes. But if we had more evidence about a timeline and certain disease presentations, it might help outcomes.
Marini:
With soft beds we almost never see decubitus ulcers any more. I'm sorry to say that, although they try, our nurses don't always turn every 2 hours, especially at night or when very busy. They find a comfortable position for the patient until the patient complains or it's time to change shifts.
Gajic:
Because of improved out-patient management in our community, I rarely see severe status asthmaticus in our ICU. But there's a population of adult cystic fibrosis patients who are on prolonged mechanical ventilation awaiting lung transplant who come with a third or fourth episode of ventilator-associated pneumonia, and they have those copious secretions and fulfill the ARDS criteria. They're taking 45 breaths a minute, and I don't know what to do with the ventilator, so we do bronchoscopy and all of that. Neuromuscular blockade does not make much sense in these patients. How should we deal with a patient who has ARDS with copious airway secretions and cystic fibrosis damaged lungs?
Marini:
To help get the secretions out you could put them in prone position. Once the secretions have disseminated, I think the risk of propagation is irrelevant. Getting the secretions out is the priority. In trying to prevent propagation, if they have one side that's involved, we incline them and use a lung-protective strategy with small tidal volumes and a generous PEEP, to try to keep the secretions in the periphery. But in most cases you're trying to evacuate the secretions, and you have to use the usual measures of positioning and suctioning, and things that contribute to propagation are irrelevant in that situation.
Branson:
John, would it ever be useful for a ventilator to have an insufflation-exsufflation system built in to help remove secretions?
Marini:
I think it would—if it worked. It could be dangerous if you don't have full recruitment. Some ventilator manufacturers are trying to incorporate a cough function. One of the problems with endotracheal tubes is that the lumen is in the center, and getting the secretions to jump the hoop and go into the external circuit is an important problem. It might make an interesting contribution to consider in-exsufflation associated with our ventilatory prescription, but there are dangers.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Introduction
- Functional Impairment in Ventilated Patients With Obstructive Disease
- Summary
- Footnotes
- References
- Hess:
- Marini:
- Hess:
- Marini:
- Branson:
- Marini:
- Hess:
- Marini:
- MacIntyre:
- Hess:
- Marini:
- Branson:
- Marini:
- MacIntyre:
- Marini:
- MacIntyre:
- Marini:
- MacIntyre:
- Marini:
- MacIntyre:
- Kallet:
- Marini:
- Kallet:
- Marini:
- Gajic:
- Marini:
- Branson:
- Marini:
- References
- Figures & Data
- Info & Metrics
- References