

Abstract
The overall goal of the cardiorespiratory system is to provide the organs and tissues of the body with an adequate supply of oxygen in relation to oxygen consumption. An understanding of the complex physiologic interactions between the respiratory and cardiac systems is essential to optimal patient management. Alterations in intrathoracic pressure are transmitted to the heart and lungs and can dramatically alter cardiovascular performance, with significant differences existing between the physiologic response of the right and left ventricles to changes in intrathoracic pressure. In terms of cardiorespiratory interactions, the clinician should titrate the mean airway pressure to optimize the balance between mean lung volume (ie, arterial oxygenation) and ventricular function (ie, global cardiac output), minimize pulmonary vascular resistance, and routinely monitor cardiorespiratory parameters closely. Oxygen delivery to all organs and tissues of the body should be optimized, but not necessarily maximized. The heart and lungs are, obviously, connected anatomically but also physiologically in a complex relationship.
- cardiorespiratory interactions
- cardiac output
- oxygen delivery
- oxygenation
- acidosis
- neonate
- pediatric
- oxygen consumption
- oxygen
- mechanical ventilation
- hypoxia
- hypoxemia
Introduction
The overall goal of the cardiorespiratory system is to provide the organs and tissues of the body with an adequate supply of oxygen in relation to oxygen consumption (V̇O2). Generally, this balance is easily maintained in the healthy person. However, cardiorespiratory dysfunction and/or abnormalities of oxygen utilization, as can occur with sepsis, inborn errors of metabolism, and some toxicities, can lead to the failure of adequate oxygen delivery to the cells of the body. The result can be metabolic acidosis, hypoxic-ischemic injury, and ultimately organ dysfunction/failure. From the perspective of the clinician, an understanding of the complex physiologic interactions between the respiratory and cardiac systems is essential for optimal patient management.
Equally important is an understanding of each patient's pathophysiology. An imbalance between oxygen delivery and V̇O2 will be managed differently depending on the specific pathophysiology. The following scenarios require differing management strategies: decreased arterial oxygen content due to ventilation/perfusion mismatching, poor cardiac output secondary to ventricular dysfunction, altered oxygen utilization related to sepsis, decreased cardiac output secondary to severe hypovolemia, and decreased arterial oxygen content related to profound anemia. Most of these conditions can be managed by increasing the arterial oxygen content and/or cardiac output. However, it should be noted that septic shock1,2 is a specialized situation characterized by mitochondrial dysfunction, impaired oxygen extraction capability, and/or abnormal tissue oxygen utilization. Thus, augmenting cardiac output in a patient with septic shock should not be expected to correct tissue hypoxia, unless there is associated cardiac dysfunction.
As oxygen delivery is a broad topic, the primary focus of this article is the effects of positive-pressure ventilation (PPV) on hemodynamics and cardiac output. The use of inotropes/vasoactive agents, intravascular fluid administration, and mechanical support devices to augment cardiac output is beyond the scope of this paper.
Mechanical Ventilation and Hemodynamics: An Overview
From both respiratory and cardiac perspectives, ventilator management for critically ill patients should be aimed at the specific needs of the individual patient, providing the highest benefit with the least risk of complications. The criteria for initiating mechanical ventilation vary according to the intended goals and pathophysiology of the clinical situation. It should be noted from the start that PPV can positively or negatively impact the cardiovascular status, although most often, no overall effect is seen due to the body's ability to compensate for changes in intrathoracic pressure.
It should also be stressed that the key parameter affecting cardiorespiratory interactions is mean airway pressure (̄Paw), which directly influences mean intrathoracic pressure. Any cardiovascular effects of the phasic change in airway pressure (ie, difference between peak inspiratory pressure and PEEP) are generally minor and must be balanced by the effects of ̄Paw. Alterations in intrathoracic pressure are transmitted to the heart and lungs and can dramatically alter cardiovascular performance. As described in more detail below, significant differences exist between the physiologic response of the right and left ventricles to changes in intrathoracic pressure.3
The discussion that follows focuses on the effects of respiratory interventions and their associated effects on mean intrathoracic pressures on the heart and pulmonary vasculature. The complex interplay between the ventilator and the right and left ventricles and pulmonary vasculature is discussed. Adjunct therapies that may affect cardiorespiratory interactions are considered as well.3–6
Cardiorespiratory Economics
Oxygen Delivery
Oxygen delivery (DO2) is the product of cardiac output and arterial oxygen content (CaO2). Oxygen content is affected by the dissolved and bound components of oxygen in the blood as well as hemoglobin (Hb).
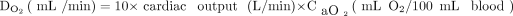 (1)
(1)
 (2) where 1.34 is the amount of oxygen (mL) carried by 1 g of hemoglobin, and 0.003 is the solubility of oxygen in plasma.
(2) where 1.34 is the amount of oxygen (mL) carried by 1 g of hemoglobin, and 0.003 is the solubility of oxygen in plasma.
In critically ill patients, especially those with severe lung injury, intrapulmonary shunt and ventilation/perfusion mismatching can result in profound hypoxemia, which can compromise arterial oxygen content and cause tissue hypoxia, especially if associated with decreased cardiac output, low hemoglobin concentration, and/or increased metabolic demand. As oxygen delivery is a function of cardiac output and arterial oxygen content, cardiac output augmentation with preload optimization, inotropic agents, vasodilators, optimization of mechanical ventilation, and, when indicated, mechanical cardiac support devices can maintain adequate oxygen delivery, even if arterial oxygen content is diminished. Thus, oxygen delivery may be augmented by increasing cardiac output, oxygen saturation, and/or hemoglobin content. The various components of oxygen delivery are displayed in Figure 1.
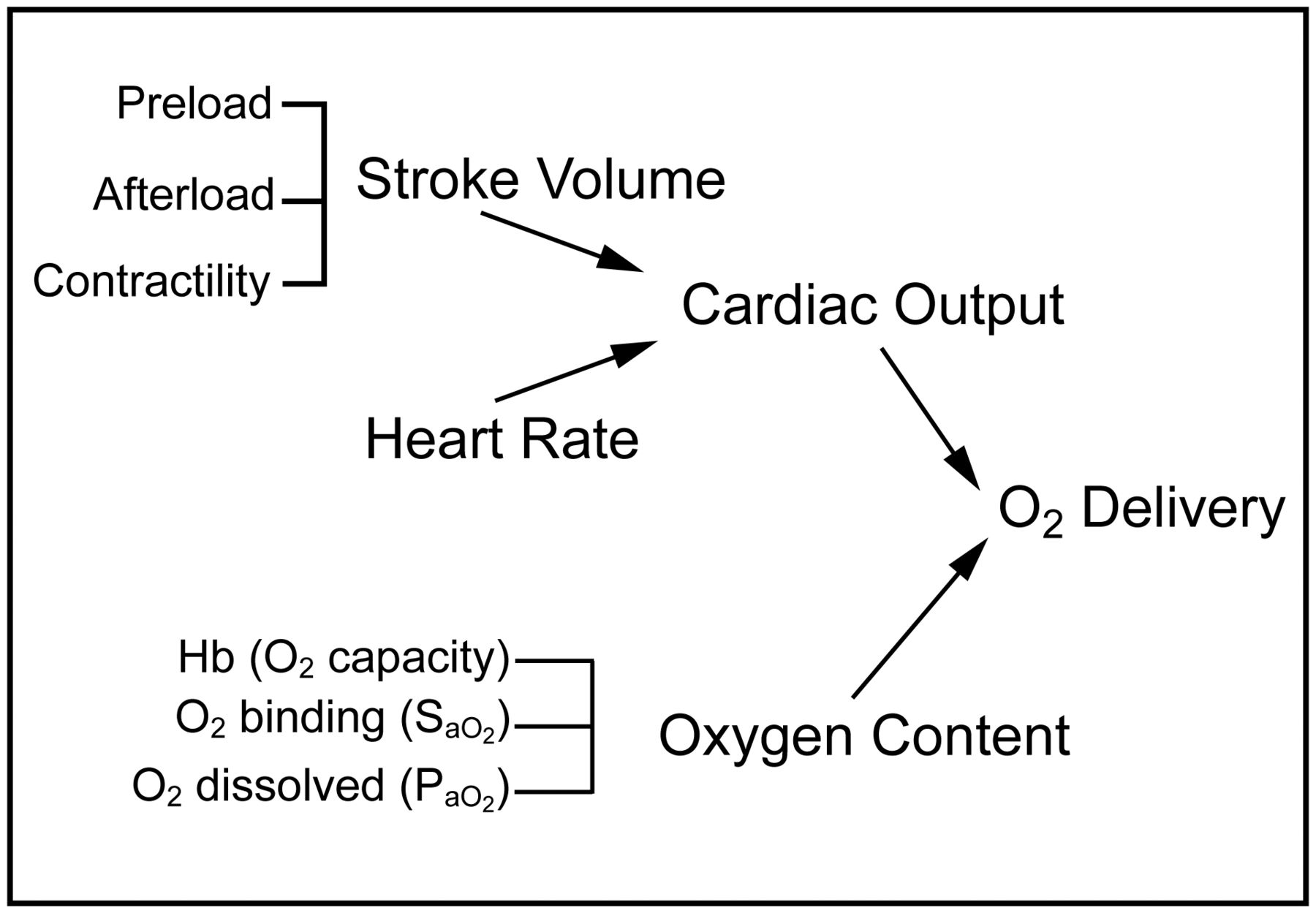
Primary determinants of oxygen delivery. Hb = hemoglobin; SaO2 = arterial oxygen saturation.
Accordingly, cardiac output, arterial oxygen content, and hemoglobin concentration are physiologically interdependent, and a decrease in one component may be balanced by a compensatory increase in another. Therefore, tissue hypoxia may not occur during hypoxemia or anemia if cardiac output is adequately increased. Clinical studies have demonstrated that cardiac output increases substantially in normal subjects during acute hypoxemia.7,8
Oxygen Consumption
In direct relation to the oxygen delivery needed to avoid end-organ injury is V̇O2, which is the product of cardiac output and the arteriovenous oxygen content difference (C(a-v)O2).
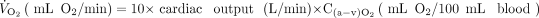 (3)
(3)
V̇O2 can be decreased by appropriately managing patient-ventilator asynchrony, providing appropriate patient work of breathing, and avoiding excessive agitation, shivering, and hyperthermia.
Effects of Mechanical Ventilation on the Right Ventricle
Physiologic differences affect the interaction of PPV on the right and left sides of the heart.3–6 The right ventricle receives blood from beyond the thorax (ie, from the superior vena cava and inferior vena cava) and pumps it within the thorax. As such, the flow of blood to the right ventricle is sensitive to alterations in mean intrathoracic pressure for several physiologic reasons.3
Systemic venous return to the right atrium is passive, with flow occurring as a result of a pressure gradient between the superior/inferior vena cava and right atrium. When the right atrial pressure is low, there is minimal resistance for venous return to the right atrium (Fig. 2). As right atrial pressure increases, resistance to systemic blood return increases, thus reducing venous return and the preload available to the right ventricle. The increased intrathoracic pressure created by PPV is transmitted to the right atrium, which, if excessive or if the patient is hypovolemic, can impede venous return and decrease right ventricular preload. This decrease in right ventricular preload impairs right ventricular performance by decreasing stroke volume and thus adversely affecting cardiac output.

Effects of mean intrathoracic pressure on systemic vascular return. Systemic venous return to the right atrium is passive, with blood flow occurring as a result of a pressure gradient between the superior/inferior vena cava and right atrium. PSV = systemic venous pressure; RAP = right atrial pressure; PPV = positive-pressure ventilation.
During spontaneous breathing (ie, negative intrathoracic pressure), right atrial pressure is low, impedance to blood flow to the right ventricle is also low, and systemic venous return is normal. Initiation of PPV (invasive or noninvasive) increases intrathoracic pressure, which is transmitted to the right heart (ie, increased right atrial pressure) and can result in a decrease in systemic venous blood return (ie, decreased right heart preload). This effect is most pronounced in situations of significant increases in mean intrathoracic pressure and/or decreases in intravascular volume (eg, septic, hemorrhagic, or hypovolemic shock). In summary, PPV increases mean intrathoracic pressure and reduces right ventricular performance by decreasing right ventricular preload.
When increases in intrathoracic pressure are needed to obtain adequate oxygenation and right ventricular output is compromised, intravascular volume administration may be indicated. As shown in Figure 3, intravascular fluid administration shifts the systemic venous return line upward and rightward. Thus, for the same right atrial pressure (mean intrathoracic pressure), systemic venous return is augmented. Although the amount of fluid required to obtain this physiologic benefit is based on the patient's intravascular status and the magnitude of the increase in ̄Paw (mean intrathoracic pressure), in general, ∼5 mL/kg is required. Subsequent fluid boluses should be administered as clinically indicated based on the pathophysiology and the individual patient's physiologic response.

Effects of preload augmentation on systemic vascular return. Intravascular fluid administration shifts the systemic venous return line upward and rightward. Thus, for the same right atrial pressure (mean intrathoracic pressure), systemic venous return is augmented. PSV = systemic venous pressure.
Although changes in intrathoracic pressure do not directly affect myocardial contractility, indirect effects can occur. Myocardial perfusion (ie, blood flow) is dependent on intrathoracic pressure, aortic pressure, and right ventricular systolic pressure. An increase in intrathoracic pressure, increase in right ventricular systolic pressure, and/or reduction in aortic pressure will reduce right ventricular myocardial blood flow. In most clinical situations, aortic pressure exceeds right ventricular and intrathoracic pressures, and right ventricular myocardial blood flow is not affected by PPV. When low aortic pressure exists, moderate-to-severe right ventricular dysfunction occurs, and/or increased intrathoracic pressure is present, these complex interactions can be clinically important. When these predisposing conditions are present, the adequacy of right ventricular myocardial perfusion should be addressed, and interventions (eg, reducing mean intrathoracic pressure, if possible, and/or increasing systemic systolic pressure) considered to optimize right ventricular myocardial perfusion and thus right ventricular function.
In addition to the primary effect of PPV on right ventricular preload and the potential secondary effect on right ventricular contractility, the effects of mechanical ventilation on pulmonary vascular resistance (PVR) and right ventricular afterload must be considered.9,10 At low lung volume, PVR is elevated due to hypoxic vasoconstriction and tortuosity of the medium and large intrapulmonary blood vessels. As lung volume increases toward functional residual capacity, PVR decreases as the larger blood vessels expand and become less tortuous. At a point significantly above functional residual capacity, increases in PVR occur due to compression of the intra-alveolar capillaries by overexpanded alveoli. As intrathoracic pressure and lung volume are altered, variable changes in PVR and right ventricular afterload occur. Subsequently, right ventricular output and global cardiac output are affected. These physiologic principles are discussed in more detail in the corresponding section below.
Effects of Ventilator Manipulations on the Left Ventricle
In contrast to the right ventricle, the left ventricle receives blood from within the thorax (ie, right ventricle via the pulmonary circulation) and pumps it outside the thorax.3 Thus, the physiologic effects of mechanical ventilation on the left ventricle are quite different from those on the right ventricle as described previously. Ventricular interdependence dictates that the left ventricle can only eject the quantity of blood it receives from the right ventricle.11 When right ventricular output is decreased (in relation to the effects of PPV or any other etiology), the blood flow to the left side of the heart (ie, left ventricular preload) decreases. Furthermore, right ventricular afterload, and thus right ventricular systolic pressure, can increase with elevation of the ̄Paw.4 An increase in right ventricular pressure can cause a conformational change in the interventricular septum and a subsequent decrease in left ventricular compliance, preload, and potentially output.
Historically, a ventilator approach used to improve left ventricular preload was thoracic augmentation of left ventricular filling.3,5 This concept describes a phasic increase in intrathoracic pressure similar to a proposed mechanism for increasing cardiac output with chest compressions during cardiopulmonary resuscitation. As the left ventricle derives its preload from intrathoracic sources, the phasic increase in intrathoracic pressure is transmitted to the pulmonary vascular capacitance, resulting in an increase in pulmonary vascular pressure above the left-sided filling pressure. As intrathoracic pressure increases, the pressure in the pulmonary vasculature further exceeds left atrial pressure, creating a pressure gradient that augments left-sided preload and cardiac output. Thus, left ventricular preload can be augmented by this thoracic augmentation physiology. However, this traditional strategy is no longer employed, as the tidal volume (VT) required to achieve a clinically pertinent increase in cardiac output can approach 15–20 mL/kg, which violates the clinical approach of low VT for lung-protective ventilation.
Although left ventricular contractility is unaffected by mechanical ventilation, left ventricular output can be dramatically altered by mean intrathoracic pressure-dependent effects on left ventricular afterload. Left ventricular afterload is dependent on left ventricular myocardial wall tension,5 which is affected by the difference between the left ventricular systolic pressure and mean intrathoracic pressure.5,6 The physiologic effects of PPV on left ventricular wall tension are complex, as the systemic arterial system has both intrathoracic and extrathoracic components. When mean intrathoracic pressure is increased, the pressure is rapidly transmitted to the intrathoracic arterial system. Left ventricular wall tension remains the same, as the left ventricular pressure and intrathoracic pressure generated are equally affected. The extrathoracic arterial system also senses this increase in arterial pressure due to the propagation of the pressure waveform into the peripheral circulation.
When an increase in mean intrathoracic pressure results in a significant increase in arterial pressure, aortic pressure will autoregulate secondary to baroreceptor stimulation.5,6 A reflexive decrease in aortic pressure occurs along with a compensatory reduction in left ventricular pressure. When aortic pressure reflexively returns to baseline, left ventricular systolic pressure decreases. as does the transmyocardial pressure gradient. Therefore, a persistent elevation in mean intrathoracic pressure decreases left ventricular wall tension (ie, decreased left ventricular afterload) as a result of aortic pressure autoregulation. These physiologic principles are shown schematically in Figure 4. On the left side of this example, the left ventricle of a spontaneously breathing patient needs to generate a transmural pressure of 120 mm Hg in response to a systemic systolic pressure of 100 mm Hg and a mean intrathoracic pressure of −20 mm Hg. When this patient is mechanically ventilated with a mean intrathoracic pressure of +20 mm Hg, the left ventricle has to generate a pressure of only 80 mm Hg to result in the same systemic systolic pressure of 100 mm Hg. Thus, an augmentation of mean intrathoracic pressure will reduce left ventricular afterload and potentially improve overall left ventricular function.

Effects of intrathoracic pressure on left ventricular (LV) afterload. An augmentation of mean intrathoracic pressure will reduce left ventricular afterload. PPV = positive-pressure ventilation; AO = aorta.
The impact of PPV and its associated beneficial effect on the left side of the heart can be an important consideration in the decision to initiate mechanical ventilation in the setting of impaired myocardial performance (eg, cardiomyopathy, myocarditis). These physiologic principles should also be considered when deciding on the timing of extubation in such a patient. As a secondary effect, a patient with cardiovascular compromise may benefit from a reduction in V̇O2 by reducing his/her work of breathing. It should be noted that the clinician must carefully weigh the potential risks and benefits of this high-risk intubation for each patient based on the individual physiology and pathophysiology. Similarly, extubation of the patient with left ventricular dysfunction/failure may lead to decompensation as left ventricular afterload and the stress on the left heart is increased. In summary, the complex interaction between intrathoracic pressure, myocardial wall tension, and aortic autoregulation leads to decreased left ventricular afterload and further augmentation of cardiac output in the setting of persistently elevated mean intrathoracic pressure, as occurs with PPV.
It should be noted that under common clinical conditions (ie, normal ventricular function), mean intrathoracic pressure is low compared with left ventricular pressure, and inspiration occurs over a very limited number of cardiac cycles. The result is only minor phasic changes in left ventricular afterload, and thus, autoregulation may not occur to a clinically relevant degree. However, if mean intrathoracic pressure is high, a sustained increase in peak intrathoracic pressure occurs over multiple cardiac cycles, and/or the left ventricle is dysfunctional, left ventricular afterload and left ventricular output can be significantly reduced.
Effects of Mechanical Ventilation on the Pulmonary Vasculature
As briefly discussed previously, right ventricular afterload is affected by lung volume (Fig. 5). When lung volume is low (ie, atelectasis, lung collapse), PVR is elevated secondary to hypoxic pulmonary vasoconstriction or the tortuous course of the large-to-medium-size pulmonary blood vessels (Fig. 5A).9,10 As lung volume increases, these larger pulmonary vessels become linear with their capacitance increasing, hypoxia improves, and PVR decreases. As lung volume continues to increase, hyperexpansion of the alveoli and compression of the pulmonary capillaries can occur, and vascular resistance increases (Fig. 5B).12 The total PVR becomes the sum of these two scenarios, and thus, PVR can be elevated at small or large lung volumes and is lowest at an ideal lung volume. Of course, the clinical challenge is determining this optimal point (Fig. 5C).

Effects of mean lung volume on pulmonary vascular resistance (PVR). Right ventricular afterload is affected by mean lung volume. A: Component of PVR related to the larger pulmonary vessels. B: Component of PVR related to the smaller pulmonary vessels. C: Total PVR.
Thus, PPV can reduce right ventricular afterload in patients with low lung volume by expanding collapsed lung units and reducing PVR. PPV can increase right ventricular afterload in conditions of pulmonary overdistention with excessive alveolar expansion and subsequent compression of the perialveolar capillaries.12
In addition to optimizing lung volume, therapy directed at reducing pulmonary hypertension can consist of increasing pH, decreasing PaCO2, increasing PaO2 and alveolar partial pressure of oxygen, and minimizing intrathoracic pressures.13–16 Increasing pH has been shown to significantly reduce PVR in a variety of studies.15,17,18 Both an increase in pH and a reduction in PaCO2 can independently reduce right ventricular afterload. In a classic report by Lyrene et al,19 the effects of alkalosis on lowering PVR were similar regardless of whether the alkalosis was metabolic or respiratory in etiology. The use of respiratory and/or metabolic alkalosis must be directed by the patient's overall clinical status and the relative risks and benefits of each approach.
In another classic study, Malik and Kidd16 demonstrated that at a buffered pH (7.4), decreases in PaCO2 did not affect PVR, whereas increases in PaCO2 increased PVR. Thus, for those patients with pulmonary hypertension, hyperventilation without a related increase in pH will not have a clinically beneficial effect. However, for those patients in whom an increase in PVR is desired (eg, mechanically ventilated infants with single-ventricle physiology and pulmonary overcirculation), an increase in PaCO2, irrelevant of a decrease in pH, will be effective.
Additionally, studies have shown that an increase in alveolar oxygen and arterial oxygen (PaO2) by increasing FIO2 can reduce PVR.15,16 Increasing the FIO2 improves PaO2 in patients without a right-to-left shunt and reduces pulmonary artery vascular resistance. Increasing inspired oxygen in patients with intracardiac shunts produces little change in PaO2; however, a reduction in PVR still results. Thus, an increase in alveolar and/or arterial oxygen content can reduce PVR.
In animal studies, increasing FIO2 has been shown to be a more potent pulmonary vasodilator in neonates than in adults.16 The use of inspired oxygen to reduce PVR has been useful in the ICU and is a common approach to assessing the reactivity of the pulmonary vascular bed in the cardiac catheterization laboratory.16
In conditions of increased afterload, a reduction in PVR will decrease right ventricular afterload and lead to an improvement of right ventricular function. These beneficial cardiorespiratory interactions may be employed, especially after congenital heart surgery and in those with pulmonary hypertension, to assist patients with right ventricular dysfunction. Beyond the use of oxygen, inhaled nitric oxide and other pulmonary vasodilators may be employed.
It should be noted that a reduction in PVR is not always beneficial, as can be seen in patients with large intracardiac or extracardiac shunts. The addition of inspired oxygen can reduce PVR and increase pulmonary blood flow. In conditions of decreased pulmonary flow, this may improve oxygen delivery by increasing arterial oxygen content. However, in clinical situations of increased pulmonary flow (eg, ventricular septal defect, large patent ductus arteriosus, and hypoplastic left heart syndrome), the associated increase in pulmonary blood flow can result in pulmonary overcirculation and pulmonary edema. For those patients with single-ventricle physiology, the resultant pulmonary overcirculation may occur at the expense of systemic output with a reduction in global oxygen delivery.
In summary, PVR (and right ventricular afterload) can be minimized when lung volume is optimal, pH is normal or increased, and PaCO2 is not increased. Additionally, pulmonary vasodilators (eg, inhaled nitric oxide, prostacyclin), whether inhaled or administered intravenously, can help to reduce PVR and potentially optimize cardiorespiratory interactions. A detailed discussion of these pharmacologic agents is beyond the scope of this article.
Effects of Ventilator Manipulations on Heart Rate
PPV generally causes minor changes in heart rate. Overdistention of the lung can result in a reflex bradycardia; however, changes in heart rate tend to be limited at the VT commonly used in clinical practice. However, ventilation with excessive VT can result in a reflex bradycardia that may become clinically important. Profound hypoxia with depressed oxygen delivery can result in bradycardia as well. Overall, the effects of PPV on heart rate tend to be less pertinent than the other physiologic changes discussed throughout this article.
Ventilator Approach for Patients With Congenital Heart Disease
Patients with congenital heart lesions represent a specialized population. The principles of cardiorespiratory interactions apply to these patients in a similar fashion as the general population; however, the effects tend to be more pronounced. Furthermore, the clinician should carefully consider the physiologic effects of mechanical ventilation on the systemic and pulmonary ventricle(s) rather than on the more traditional right- and left-sided principles. In those patients with single-ventricle physiology, the ventricle can have systemic and pulmonary physiologic components, which will likely differ in the preoperative state compared with the palliated postoperative state. Because of the complex cardiorespiratory interactions that occur and the diversity of the conditions treated, a single standardized approach is not possible. Respiratory strategies should be designed to address the specific pathophysiologic condition present in each patient.
Increased Oxygen Consumption
Although the clinician has multiple approaches for optimizing oxygen delivery, the options for minimizing/optimizing V̇O2 are more limited. In the situation in which V̇O2 is excessive in relation to oxygen delivery, the clinician should consider treating patient-ventilator dyssynchrony, titrating ventilator support to provide an appropriate patient work of breathing, and avoiding excessive patient agitation, shivering, and hyperthermia.20,21
Patient-ventilator dyssynchrony22–24 can lead to excessive V̇O2 by the respiratory muscles. Dyssynchrony can be flow-related or trigger-related and occurs when spontaneous inspiratory effort is out of phase with the ventilator-delivered breaths.22–24 When dyssynchrony is present, primary hypoxemia due to ventilation/perfusion mismatching, mucous plugging, pneumothorax, and reactive airway disease must be eliminated as the etiology. When these causes are eliminated, altering the mode (ie, inspiratory flow pattern), improving the trigger sensitivity, or increasing the support provided by the ventilator may improve patient-ventilator synchrony. Improving patient-ventilator synchrony can reduce V̇O2, especially for infants and small children.
Oxygen Delivery
Although an important clinical goal is to maintain an optimal balance between oxygen delivery and V̇O2 for the critically ill patient, the clinician is often in a situation in which this balance cannot be quantified. Pulmonary artery/cardiac output catheters are less commonly used than in the past, especially for infants and children. Thus, the clinician is often left with surrogate end points.
Poor oxygen delivery (or a mismatch between delivery and consumption) can be seen clinically by decreased renal function, abnormal mental status, and poor right ventricular function. It must be noted that these clinical assessments can be altered by the use of diuretics and pharmacologic sedation. Additionally, right heart dysfunction as a result of poor oxygen delivery is a late marker of a problem.
Beyond clinical assessment, the clinician can investigate an imbalance between oxygen delivery and V̇O2 by evaluating laboratory markers of metabolic acidosis as an indicator of global oxygen debt. Such studies to confirm that tissue oxygen supply is adequately maintained include mixed venous oxygen saturation (Sv̄O2), blood lactate, base deficit, and arterial pH.
Sv̄O2 can be measured intermittently via repeated blood gas analysis or continuously via a fiberoptic catheter.18,25 Sv̄O2 is probably the best single indicator of the adequacy of oxygen transport, as it represents the amount of oxygen remaining in the systemic venous blood after blood passes through the organs and tissues throughout the body.26 Sv̄O2 reflects the balance between oxygen supply and demand and can be a surrogate for cardiac output as a target for goal-oriented hemodynamic therapy.27 It should be noted that the oxygen extraction ratio may be preferable to Sv̄O2 as an indicator of global tissue hypoxia because arterial hypoxemia reduces Sv̄O2 without necessarily indicating oxygen debt (ie, the oxygen extraction ratio can be normal, whereas arterial oxygen saturation [SaO2] and Sv̄O2 are proportionally reduced).
It must be stressed that all markers of oxygen delivery are global indicators. Regional areas of tissue/organ hypoxia may exist. Specific examples include sepsis, necrotizing enterocolitis in the neonate, acute kidney injury in the critically ill patient with shock, and intracranial injury. Alterations of microcirculatory blood flow reduce the sensitivity of global markers of tissue hypoxia to detect regional abnormalities of tissue oxygenation.26,28 In sepsis, tissue hypoxia is largely due to impaired oxygen utilization, although Sv̄O2 may be normal or high.29,30
Noninvasive monitoring of cerebral and splanchnic oxygen saturation is being increasingly advocated as a reliable assessment of oxygen delivery to the vital organs of the body.31,32 However, it must be noted that the clinical reliability of the use of near-infrared spectroscopy to monitor oxygen delivery remains controversial.33
Tissue oxygen delivery is determined not only by SaO2 but also by the patient's hemoglobin content, cardiac output, oxygen affinity of hemoglobin, oxygen extraction ratio, and metabolic demands. SaO2 is not a sensitive index of tissue oxygenation/oxygen delivery. It must be kept in mind that, to date, no randomized trial has assessed the relationship between SaO2 and tissue oxygenation in critically ill patients.
As the clinical goal is to provide the tissues and organs of the body with an appropriate supply of oxygen, one must be aware that providing supranormal oxygen delivery is not necessarily beneficial. Randomized controlled trials have demonstrated that maintaining supranormal oxygen delivery does not improve survival and may be detrimental.27,34–36 However, other clinical trials have reported favorable outcomes with supranormal oxygen delivery.37–39 An optimal approach may be early goal-directed therapy, which has been shown to reduce mortality for patients with severe sepsis and septic shock.40 This approach involves adjustments of cardiac preload, afterload, and contractility to balance oxygen delivery with V̇O2. The physiologic rationale for an oxygen delivery early goal-directed therapy approach is to augment systemic oxygen delivery to alleviate tissue hypoxia, which can progress to multi-organ dysfunction/failure.
Despite the conflicting data, a common clinical approach is to augment oxygen delivery when the patient is on the oxygen-dependent portion of the tissue oxygenation-oxygen delivery portion of the curve (Fig. 6), but not augment oxygen delivery once the patient is on the flat portion of the curve (ie, the point between which tissue oxygenation is not oxygen delivery-dependent). The elbow of this curve is often noted as the anaerobic threshold.

Effects of oxygen delivery on tissue oxygenation. The dotted vertical line represents the anaerobic threshold.
Conclusions
Tissue and organ oxygen delivery, but not necessarily PaO2 and SaO2, must be optimized in relation to V̇O2. This fine balance varies between patients based on the individual's pathophysiology, as well as within a given patient over time as his/her clinical condition changes. The bottom line is that oxygen delivery to all organs and tissues of the body must be optimized (but not necessarily maximized). In terms of cardiorespiratory interactions, the clinician should titrate the ̄Paw to optimize the balance between mean lung volume (ie, arterial oxygenation) and ventricular function (ie, global cardiac output), minimize PVR, and routinely monitor cardiorespiratory parameters closely. The heart and lungs are, obviously, connected anatomically but also physiologically in a complex relationship.
Footnotes
- Correspondence: Ira M Cheifetz MD FAARC, Pediatric Critical Care Medicine, Duke Children's Hospital, Duke University Medical Center, Box 3046, Durham, NC 27710. E-mail: ira.cheifetz{at}duke.edu.
Dr Cheifetz has disclosed no conflicts of interest.
Dr Cheifetz presented a version of this paper at the New Horizons in Respiratory Care Symposium: Back to the Basics: Respiratory Physiology in Critically Ill Patients of the AARC Congress 2013, held November 16–19, 2013, in Anaheim, California.
- Copyright © 2014 by Daedalus Enterprises
References





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Introduction
- Mechanical Ventilation and Hemodynamics: An Overview
- Cardiorespiratory Economics
- Effects of Mechanical Ventilation on the Right Ventricle
- Effects of Ventilator Manipulations on the Left Ventricle
- Effects of Mechanical Ventilation on the Pulmonary Vasculature
- Effects of Ventilator Manipulations on Heart Rate
- Ventilator Approach for Patients With Congenital Heart Disease
- Increased Oxygen Consumption
- Oxygen Delivery
- Conclusions
- Footnotes
- References
- Figures & Data
- Info & Metrics
- References