

Abstract
Congenital parenchymal lung malformations have an estimated incidence at 1:25,000–1:35,000 births. We present a case of this rare congenital abnormality in a 38 year-old male, review the current literature with discussion of proposed causes, malignant potential, and management strategies. A 38-year-old white male presented with a 4-day history of chronic stable hemoptysis. Social history was notable for a 50-pack-year active smoking history and remote heavy alcohol consumption. Physical examination was normal. Chest radiograph revealed an ill defined right lower lobe infiltrate. Chest computed tomography demonstrated an irregular, thin-walled, cystic lesion with adjacent nodularity and calcifications. The patient received a right lower lobectomy. Pathologic specimen demonstrated a 10-cm, mostly thin-walled cyst with features suggestive of a congenital cyst adenomatoid malformation and areas of adenocarcinoma (mixed subtype with acinar and bronchioloalveolar patterns). Congenital cyst adenomatoid malformations have recently been renamed as congenital pulmonary airway malformations and are the most common type of congenital parenchymal lung malformations. Individuals typically present with recurrent pulmonary infections, pneumothorax, or hemoptysis. The development is controversial but believed to be a result of arrested development of the fetal bronchial tree during the sixth and seventh week of fetal development. Defects in thyroid transcription factor 1 have also been proposed. With the increasing use and image resolution of ultrasound in modern obstetric practice, congenital pulmonary airway malformations rarely go undetected into adulthood. Management remains controversial; however, most authors agree with early surgical excision.
- congenital cystic adenomatoid malformation
- CCAM
- congenital lobar hyperinflation
- bronchogenic cyst
- lobar sequestration
- hemoptysis
Introduction
Congenital parenchymal lung malformations are uncommon yet well described fetal anomalies, primarily identified in the pediatric population. They incorporate a spectrum of interrelated abnormalities that include congenital lobar hyperinflation, bronchogenic cyst, congenital cystic adenomatoid malformation (CCAM), and lobar sequestration.1 We present a case of hemoptysis in a 38-year-old male with CCAM and review the literature relating to the presentation of this congenital lung malformation in adults.
Case Report
A 38-year-old white male presented with 4 days of acute onset, low volume, hemoptysis. He denied infectious, constitutional, or other respiratory symptoms. Social history was notable for a 50 pack-year smoking history. Physical examination was normal. Chest radiograph revealed an ill defined right lower lobe infiltrate (Figs. 1 and 2). Computed tomography of the chest demonstrated an irregular, thin walled, cystic lesion with adjacent nodularity and minimal calcification (Fig. 3). Bronchoscopy revealed airway distortion, without endobronchial abnormality, preventing examination of the right lower lobe airways below the superior segment take-off. Bronchial washings for cytology and bacterial, fungal, and mycobacterial cultures were negative. Due to his persistent hemoptysis and concerns for possible future infectious complications, the patient underwent a thoracotomy and right lower lobectomy (Fig. 4). Intraoperative course was complicated by multiple pleural adhesions, requiring extensive dissection and placement of surgicel to control bleeding at the costovertebral junction. The patient's postoperative course was complicated by back pain and lower extremity neurologic findings, prompting emergent decompressive laminectomy and removal of surgicel expansion into the spinal canal, with subsequent cord compression. Pathologic specimen demonstrated a 10 cm, mostly thin walled cyst with features suggestive of a CCAM and areas of adenocarcinoma (mixed subtype with acinar and bronchioloalveolar patterns) (Fig. 5). Microscopic evaluation demonstrated foci of fibrosis lining the cavity, with pseudostratified columnar epithelium that transitions to adenomatous disorganized tissue consistent with a type 1 CCAM and associated adenocarcinoma (see Figs. 5 and 6). Immunohistological staining was positive for thyroid transcription factor 1 (TTF-1), consistent with the diagnosis of a primary lung adenocarcinoma.

Posterior/anterior chest x-ray demonstrating an ill defined reticular and interstitial opacity that overlies the right middle and lower lobes.

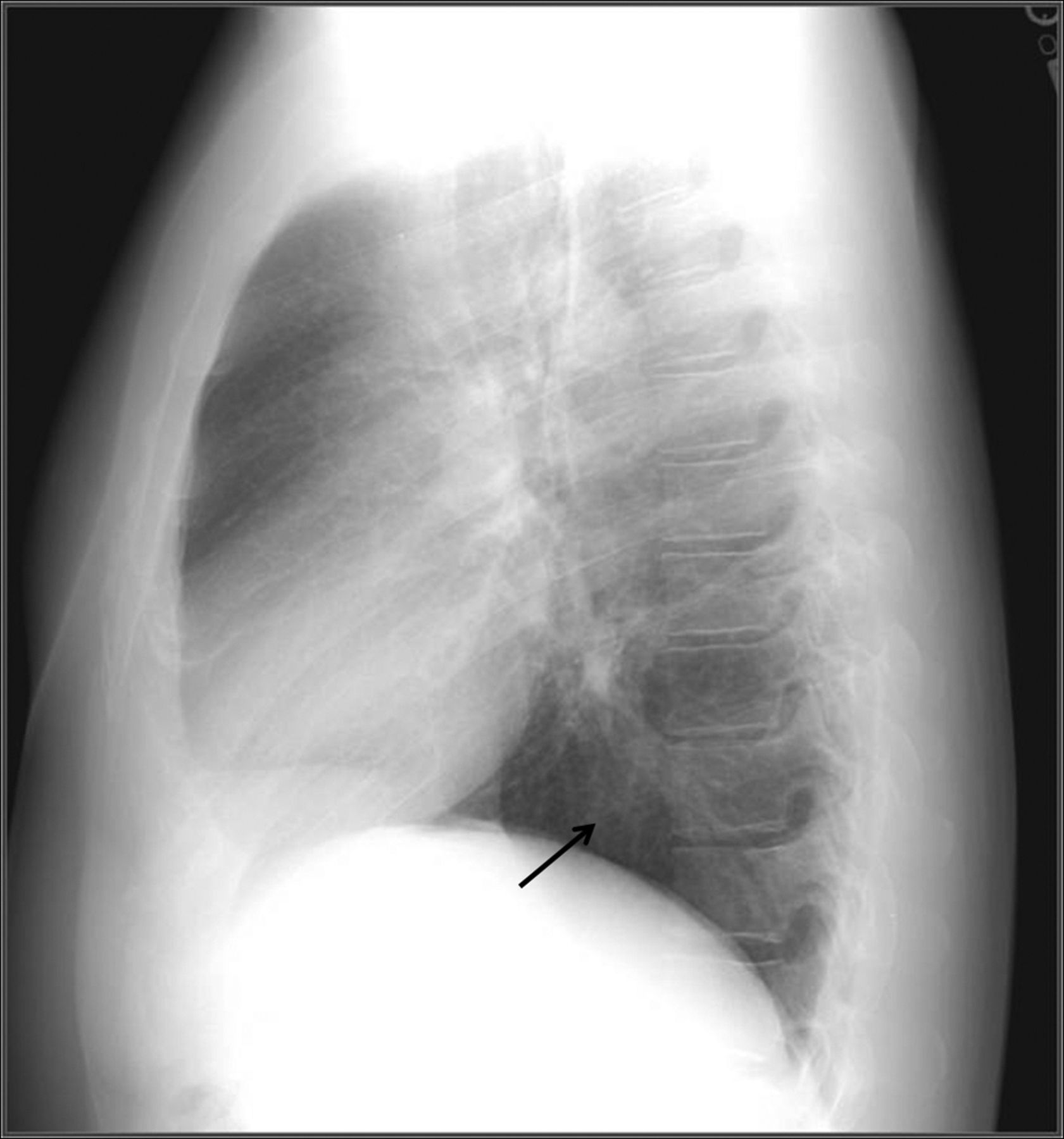
Lateral chest x-ray demonstrating reticular and interstitial opacity in the right lower lobe.

Computed tomography of the chest demonstrating an irregular, cystic lesion and right lower lobe with nodular characteristics in the superior, posterior aspects. There is adjacent atelectasis, scarring , nodules, and hyperinflation. Adjacent ground-glass may be due to recent hemoptysis or inflammation.
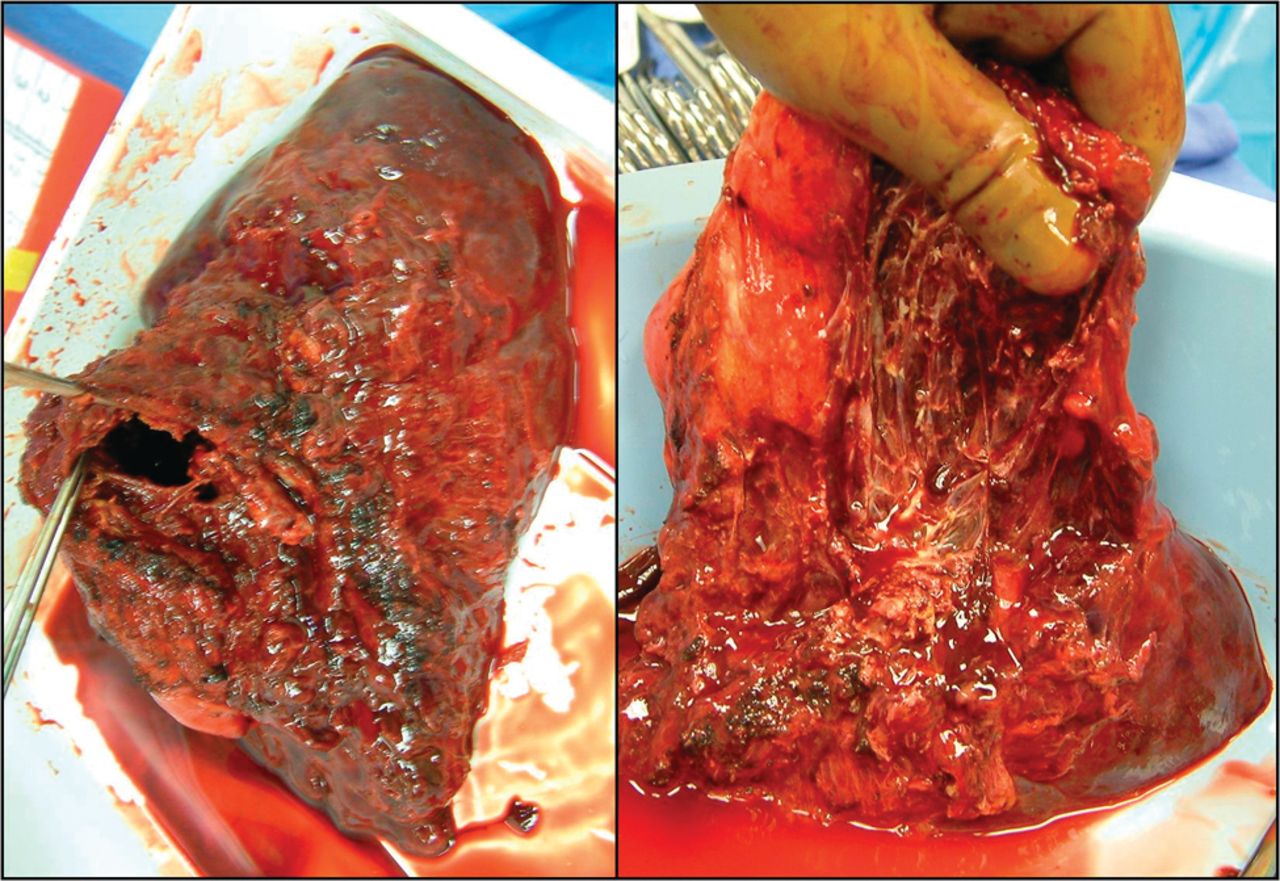
Pathologic specimen of the right lower lobe demonstrating a cavitary lesion with evidence of chronic pleural inflammation and adhesions.

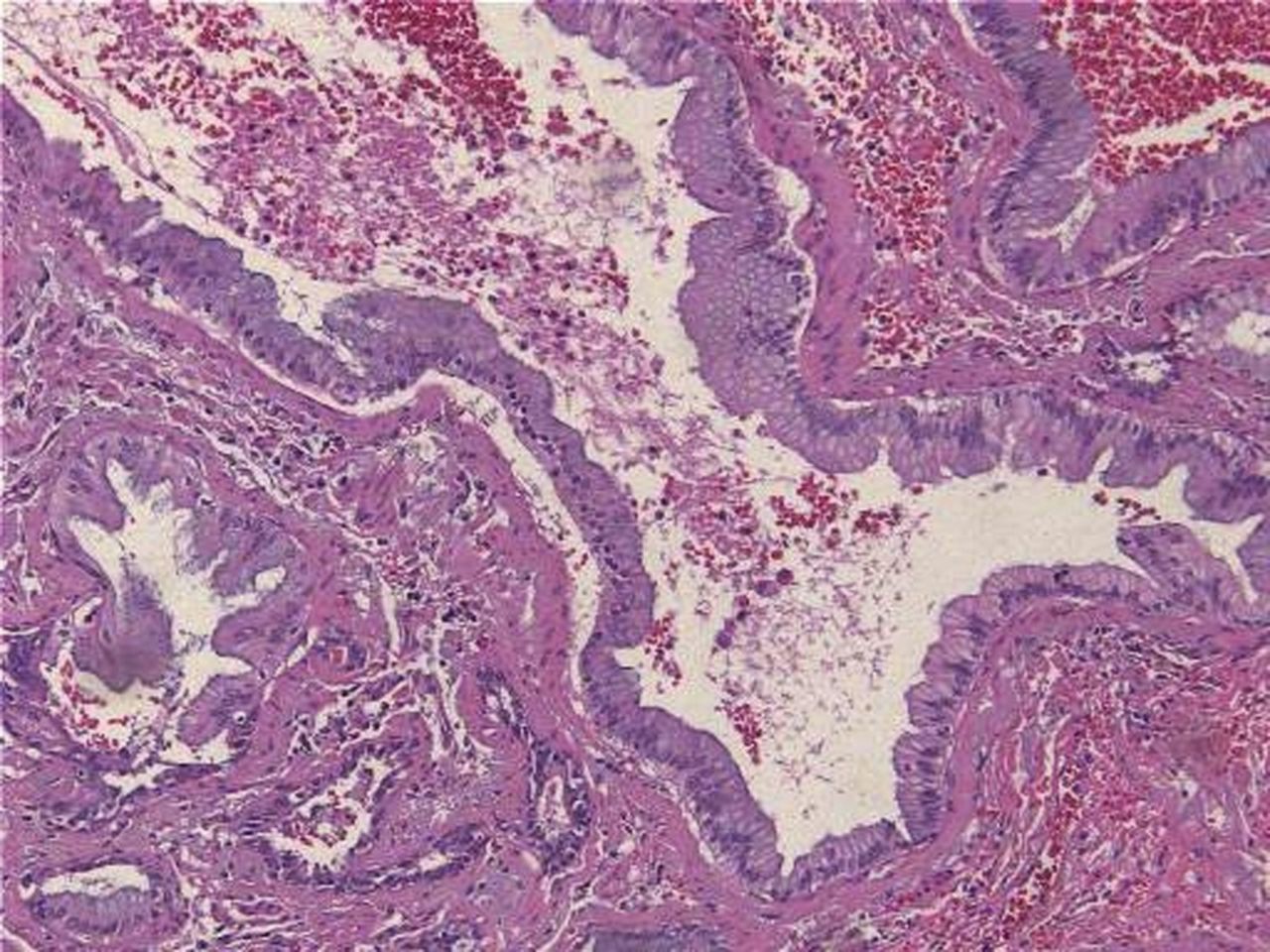
Bronchoalveolar carcinoma lining normal alveolar structure.

Thick fibrous connective tissue with islands of cuboidal and pseudocolumnar epithelium.
Discussion
CCAMs were first identified in 1949 by Ch'in and Tang as distinct and rare lesions that occurred in stillborn or premature newborns with anasarca.2 CCAMs are the most common form of congenital parenchymal lung malformations and have an estimated incidence of 1:25,000–1:35,000 births.3 The differential diagnosis includes bronchogenic cyst, pulmonary sequestration, congenital diaphragmatic hernia, and congenital lobar emphysema. CCAMs were previously categorized according to the Stocker classifications 1, 2, and 3.4 More recently this classification was expanded and the name changed to congenital pulmonary airway malformation (CPAM), reflecting the fact that not all of these lesions are cystic (Table 1).1 The staging of CPAMs from type 0 to type 4 malformations reflects the progression of developmental abnormalities through the tracheobronchial tree, from large airways to bronchioles and alveoli.
Expanded Stocker Classification for Congenital Pulmonary Airway Malformations
The pathophysiology of CPAM development is controversial. Some authors believe they result from arrested development of localized portions of the bronchial tree during the sixth and seventh week of fetal development. Others believe they are hamartomatous lesions of the bronchial tree.5,6 TTF1 is expressed solely in lung, thyroid, and certain portions of the brain. It regulates lung epithelial differentiation and plays a central role in lung development, and is used in immunohistochemical staining of malignant lesions to help differentiate primary lung cancer from metastatic.7 TTF-1 is found in normal lung tissue, but expression is solely found in bronchial and alveolar epithelial cells.8 Disruption of TTF-1 by either mutation or deletion has also been postulated to contribute to the development of CPAMs.9
CPAMs are usually identified during routine obstetric care due to the increased modern use of ultrasound, and children with this abnormality most commonly present with respiratory distress and recurrent pulmonary infections.10 The fetal lesions can remain stable in size, enlarge, or be associated with polyhydramnios or hydrops fetalis, which carries with it a poor prognosis.11,12 CPAMs account for 42% of pediatric lung resections, with 86% identified by age 5.1,7 To date, there have been 42 reported cases (Table 2) of CPAM presenting in adulthood (age > 17 y). In these cases there was near equal distribution between males (no. = 21) and females (no. = 21).12–24 The mean age on presentation was approximately 36 years old (range 17–65 years old). Adult CPAMs are most common in the lower lobe (no. = 30), with slight right (no. = 22) versus left (no. = 18) predominance. The most common presenting symptom in adults is recurrent pulmonary infections (no. = 21). Contrary to recent case reports on this subject, our review of the literature identified asymptomatic radiographic abnormalities as the second most common presentation (no. = 10, 24%) of CPAM in adults.14 Other complications include pneumothorax (no. = 5), hemoptysis (no. = 3), fever (no. = 1), dyspnea (no. = 1), and malignant transformation.3 The exact incidence of malignant transformation with CPAMs is unknown but suggested to be approximately 1%.3 A large case series in a pediatric population found a 4–9% association between CPAMs and lung tumors.25
Reported Cases of Congenital Pulmonary Airway Malformation Presenting in Adulthood
Type 1 CPAMs have mucogenic epithelium, which is hypothesized likely to be the cause of neoplastic transformation.26 Bronchoalveolar cell carcinoma (BAC) is the most common form of malignant cell type associated with CPAM in adults, which can be challenging to identify using current available imaging modalities and non-surgical biopsy sampling.25 The vast majority of authors therefore recommend surgical excision of these lesions for definitive treatment, due to the risk of malignant transformation and recurrent pulmonary infection, the latter of which typically responds poorly to medical therapy. This recommendation remains controversial, as some authors argue that the risk of malignant transformation is overstated.25 There have been no systematic reviews published comparing conservative management to surgical resection, and, given the high incidence of asymptomatic presentation that our review has identified, there may be a tendency to consider serial follow-up only in these individuals. It is worth noting one published case report that identified a 6-year-old male with a type 1 CPAM and biopsy proven BAC. He was managed conservatively over a 15 year period and at age 21 he was diagnosed with metastatic adenocarcinoma.27
Although the number of adult cases of CPAMs and associated lung cancer are small, most of the literature to date support that there is a relationship between the two. Surgical resection provides definitive management of this rare condition and its high rate of complications, and carries with it a low risk in this otherwise healthy population. Despite the complicated postoperative course in this patient, surgery remains the logical treatment strategy of choice in these authors' opinion. The complication witnessed in this case is rare but reported. Given the tendency for recurrent, chronic infection and inflammation found in association with CPAMs, a potential complicated intraoperative dissection should be anticipated as part of preoperative planning. Given the risk of continued expansion found in our case, surgicel should not be used in and around the costovertebral junction.
Footnotes
- Correspondence: Ryan McDonough DO CPT MC USA, Department of Medicine, Madigan Army Medical Center, Building 9040, Fitzsimmons Drive, Tacoma WA 98335. E-mail: ryan.j.mcdonough{at}us.army.mil.
-
The views expressed in this paper are those of the authors and do not reflect the official policy or position of the Department of the Army, Department of Defense, or the United States Government.
-
The authors have disclosed no conflicts of interest.
- © 2012 by Daedalus Enterprises Inc.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}