

Abstract
Carbon monoxide (CO) is usually recognized as a toxic gas that can be used to assess lung function in the pulmonary function laboratory. The toxicity of CO relates to its high affinity for hemoglobin and other heme molecules, producing carboxyhemoglobin (HbCO). Despite that blood HbCO levels are commonly measured in patients with CO poisoning, the clinical presentation often does not correlate with the HbCO level, and clinical improvement in the patient's condition does not correlate with HbCO clearance. In patients with CO poisoning, administration of 100% O2 is standard practice. If available, hyperbaric O2 can be used, although this is controversial. Measurement of exhaled CO might be useful to estimate HbCO, such as in smoking cessation programs, but assessment of HbCO using pulse oximetry can be misleading. Endogenous CO is generated as the result of heme oxygenase activity. It is becoming increasingly recognized that the results of heme oxygenase activity, specifically CO production, might have important physiologic functions. These include effects on vascular function, inflammation, apoptosis, cell proliferation, and signaling pathways. Given the abundance of basic science supporting a therapeutic role for CO, clinical trials are exploring this potential.
Introduction
Carbon monoxide (CO) is a colorless, odorless, and tasteless gas that is slightly less dense than air (molecular weight of 28 g/mol). It consists of one carbon atom and one oxygen atom, connected by a triple bond. It is toxic at low inhaled concentrations, and lethal at 1%. CO concentrations are usually expressed as parts per million (ppm); 1% is 10,000 ppm. CO is produced by normal metabolism at a rate of 0.4 mL/h.1 For many years, CO has been used in the pulmonary function laboratory to assess diffusing capacity. It has recently become increasingly recognized that metabolically produced CO might have important biologic benefits, and might even have therapeutic potential. In this article, I review the harms and benefits of inhaled and metabolically produced CO.
CO Toxicity
Carboxyhemoglobin
CO binds with high affinity to many ferrous heme-containing proteins. The most important of these affinities is that for hemoglobin (Hb), with which CO binds to produce carboxyhemoglobin (HbCO). Hemoglobin has a 218-fold greater affinity for CO than for oxygen.2 Exposure to CO levels as low as 10 ppm can lead to HbCO levels of 2%.3 HbCO >2–3% in nonsmokers and 10% or greater in smokers is considered abnormal.4
The adverse effects of HbCO are 2-fold. First, CO displaces O2 from Hb, thus reducing oxyhemoglobin saturation (HbO2). For example, with a HbCO of 20%, the highest possible HbO2 is 80%. This results in a reduced oxygen content and anemic hypoxia. Second, HbCO results in a left shift of the oxyhemoglobin dissociation curve, resulting in an increased affinity of Hb for O2. Thus, CO causes hypoxia by decreasing oxygen carrying capacity and also by decreasing O2 release to tissues (Fig. 1).4–8

A. CO competitively binds to Hb with oxygen, reducing carrying capacity. B: CO binding to Hb, similar to oxygen, stabilizes the relaxed R-state of Hb, increasing other sites' affinity for oxygen binding, reducing oxygen delivery by the remaining, non-CO-bound Hb (shifting the oxygen dissociation curve to the left). From Reference 8, with permission.
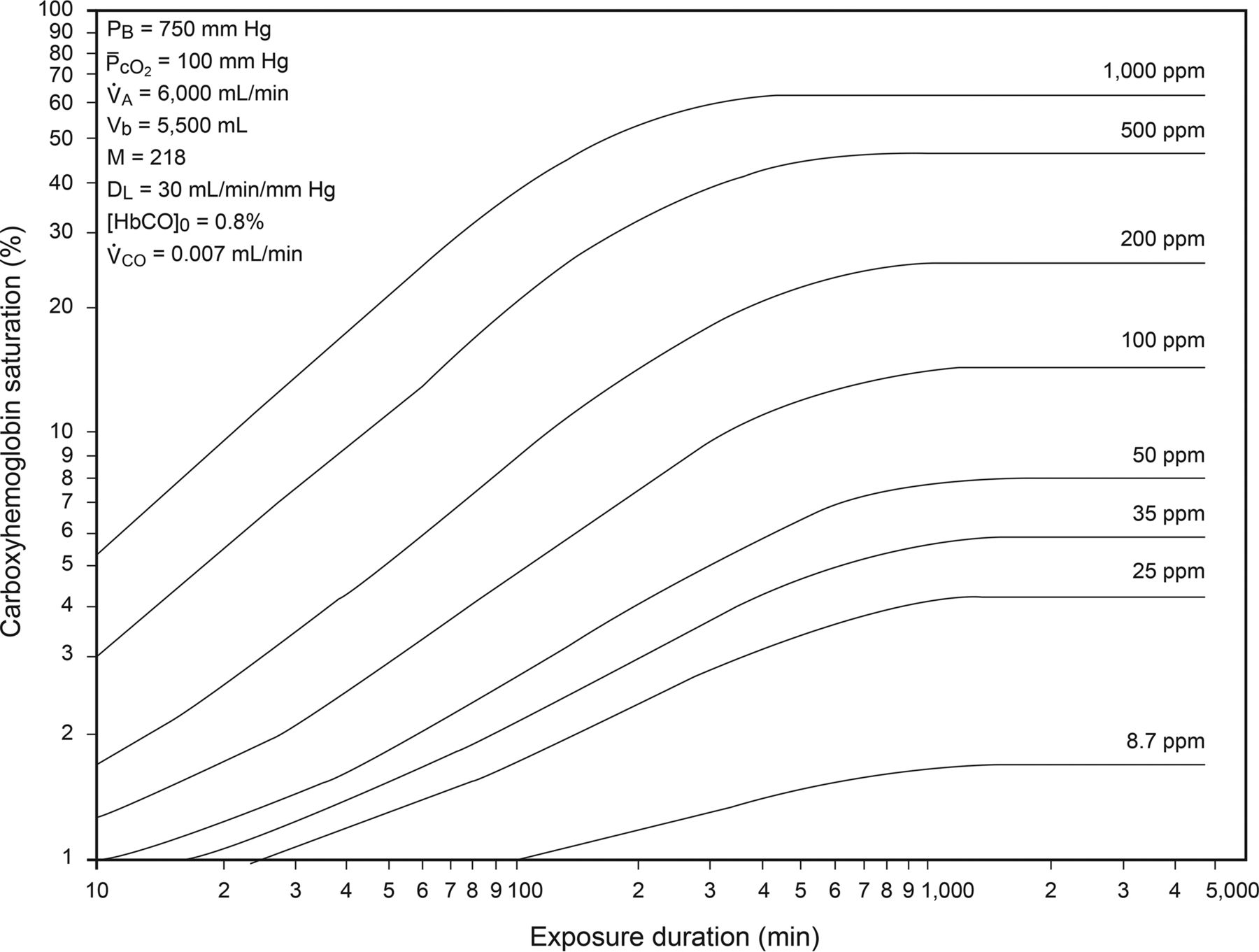
The Coburn-Forster-Kane (CFK) equation describes the kinetics of HbCO during CO exposure (Fig. 2).1 The time course of HbCO formation is determined by inhaled CO concentration, barometric pressure, PO2, alveolar ventilation, blood volume, the affinity of hemoglobin for CO, and diffusing capacity. Note that exposure for <1 h to 1,000 ppm (0.1%) CO results in an HbCO of 20%. Also note that with low-level exposure (for example, 25 ppm), the HbCO would not be expected to exceed 5%.

Carboxyhemoglobin levels as a function of exposure time and CO concentration. PB: barometric pressure; ̄PcCO2: mean PO2 in pulmonary capillaries; V̇A: alveolar ventilation; Vb: blood volume; M: ratio of the affinity of hemoglobin for CO compared to that for O2; DL: carbon monoxide diffusing capacity; HbCO0: rate of endogenous CO production; V̇CO, rate of endogenous CO production. From Reference 1, with permission.
The toxicity of CO is due to mechanisms beyond the hypoxia resulting from HbCO. CO poisoning causes direct cellular changes involving immunological or inflammatory damage by a variety of mechanisms.4 These include binding to intracellular proteins (myoglobin, cytochrome a,a3), NO generation leading to peroxynitrite production, lipid peroxidation by neutrophils, mitochondrial oxidative stress, apoptosis, immune-mediated injury, and delayed inflammation. These toxic effects of CO explain the observation that the clinical presentation (Table 1) often does not correlate with the blood HbCO level, and clinical improvement in the patient's condition does not correlate with HbCO clearance.9–11
Symptoms of CO Poisoning.
Measurement of Carboxyhemoglobin
The clinical diagnosis of acute CO poisoning requires measurement of HbCO. It is important to appreciate that a blood gas analyzer without a CO-oximeter calculates oxygen saturation assuming that HbCO is absent. Thus, for a patient with a PaO2 of 200 mm Hg and a HbCO of 20%, the blood gas analyzer will report an oxygen saturation of 100%, when in reality it cannot be greater than 80%. A CO-oximeter uses multiple wavelengths of light to measure the blood concentrations of HbO2, HbCO, and methemoglobin. CO-oximetry is the accepted standard for assessment of HbCO. HbCO concentrations are similar for arterial and venous blood.12
Inaccuracy of traditional 2-wavelength SpO2 measurement in the presence of HbCO has been long appreciated. An elevated HbCO falsely elevates SpO2, usually by an amount less than the HbCO, but this results in an extreme overestimation of HbO2. The difference between SpO2 and HbO2 due to HbCO is called the pulse oximetry gap.13 Pulse oximetry technology that uses 7 wavelengths of light is now available, instead of the usual 2 wavelengths, and is able to measure 2 species of human hemoglobin, including carboxyhemoglobin (SpCO). There have been a number of published studies assessing this technology. Common to these studies is a finding of relatively low bias but poor precision when comparing SpCO and HbCO. It is likely that the low bias is due to erroneous results occurring equally above and below the true value, which cancel each other. There may be too much error in SpCO measurement to allow its use for clinical decision making, so patients considered to be at risk for carboxyhemoglobinemia should have confirmatory blood levels measured.13 An important issue to understand is that pulse oximeters that measure SpCO use the conventional 2-wavelength algorithm to determine SpO2. Thus, when there are significant levels of HbCO, SpO2 is not a reliable method to assess arterial blood oxygen saturation.
The CFK equation1 suggests that the HbCO might be estimated if CO exposure is known. It has been known for many years that HbCO can be estimated from the CO concentration in the exhaled breath.14–16 Commercially available devices are available to measure exhaled CO.17 Stewart and colleagues14 derived the theoretical relationship between exhaled (alveolar) CO and HbCO, tested the relationship among firefighters, and reported a close relationship between the theoretical and actual values for HbCO. Wald et al15 reported a strong correlation between exhaled CO and HbCO, with the exhaled CO (ppm) roughly 5 times the HbCO. Jarvis et al16 reported that HbCO can be approximated by dividing the exhaled CO (ppm) by 6. More recently, Andersson and Moller18 evaluated the relationship between exhaled CO and HbCO. They found that the Jarvis equation,16 which was incorporated into the device they used, underestimated the HbCO measured on blood samples. However, they derived an equation that more closely estimated HbCO from exhaled CO. Hullin and colleagues19 found that blood HbCO was highly correlated with the severity of CO poisoning, whereas exhaled CO was not. They also noted that the measurement technique for exhaled CO was difficult for some subjects, which might have affected the results.
CO Exposure
Short-term exposure to inhaled CO seems to be well tolerated in otherwise normal individuals. Stewart et al20 exposed healthy volunteers to 100 ppm CO for 8 h, resulting in HbCO levels of 11–13%, with no adverse effects in time estimation, steadiness, manual dexterity, electroencephalography, and evoked potentials. Zevin et al21 exposed healthy volunteers to 1,200–1,500 ppm CO for 10 min, repeated every 45 min for 16 h/d for 7 d. Mean HbCO was 5 ± 1%, and no adverse events were reported. Ren et al22 exposed 11 normal individuals to CO, aiming to maintain a HbCO at 10% for 8 h; the actual HbCO was 9.1–10.5%. No adverse effects were reported.
CO exposure limits in the workplace have been set by the Occupational Safety and Health Administration (OSHA) and the National Institute for Occupational Safety and Health (NIOSH) (https://www.osha.gov/dsg/annotated-pels/tablez-1.html). OSHA set a permissible exposure limit for CO at 50 ppm time-weighted-average (TWA) over 8 h, 25 ppm short-term exposure limit, and 200 ppm ceiling. NIOSH set a recommended CO exposure limit of 35 ppm 8 h TWA.
CO detectors should be used in the home to alert exposure, and this is mandated in some states. It is interesting to note that the exposure limits for a home CO alarm are more generous than OSHA and NIOSH limits (http://www.carbonmonoxidedetectorhq.com/carbon-monoxide-levels-standards/). However, the CFK equation1 suggests that an exposure of 400 ppm for 15 min will result in a HbCO of < 5%. Home CO detectors (UL 2034 required alarm points) are:
0–29 ppm: The detector must remain silent. If it has a digital display, it must show a default zero reading. It may display the actual reading only if the user presses a button.
30–69 ppm: If the CO level remains in this range for 30 d, the audible alarm may sound. If a digital display is present, it should show the actual CO level as long as it is 30 ppm or higher.
70–149 ppm: If the CO level remains in this range for 1 to 4 h, the alarm must sound.
150–399 ppm: The alarm must sound if the CO level remains in this range for 10 to 50 min.
400 ppm and higher: The alarm must sound if and only if the CO level remains at this level for 4 min and for no more than 15 min.
Epidemiology of CO Poisoning
CO poisoning is not a trivial occurrence in the United States. It is responsible for hundreds of deaths and thousands of emergency department visits every year. Accidental exposures (eg, gasoline engine in an enclosed space) account for the majority of nonfatal poisonings, and intentional exposures (eg, suicide attempts) account for the majority of deaths.23 Interestingly, Hampson reported that, in the United States, deaths by CO poisoning decreased from 1,967 in 1999 to 1,319 in 2014, and death rates fell as well. Accidental poisoning accounted for 13% fewer deaths in 2014 than in 1999, and the number of intentional deaths decreased by 47% over the same period.23
Hampson23 found that the rate of decline in age-adjusted death rates was similar for the 19 states that required residential CO alarms and those that did not. It is possible that CO alarms are being installed regularly even in states without legislation requiring them, but this is not known. CO alarms are probably effective at reducing mortality. Their use should be encouraged,24,25 and there is a positive cost-benefit for their use.26 Interestingly, smoke alarms seem to be used more commonly than CO alarms.27 The most common reasons for not using CO alarms seems to be related to lack of knowledge about the devices and poor motivation. CO sensors have a limited lifespan, and modern devices emit a signal to prompt replacement.23
Treatment of CO Poisoning
The half-life of HbCO breathing normobaric room air (21% O2 at 1 atm) is 320 min.28 When breathing 100% O2 at 1 atm, the half-life of HbCO at 1 atm decreases to 74 min.29 Thus, for a patient with a HbCO of 20%, after more than 5 h breathing room air, the HbCO will have decreased to about 10%. However, if breathing 100% O2, the HbCO will decrease from 20% to 10% in a bit more than an hour. This establishes the clinical recommendation that 100% O2 should be administered to all patients with suspected CO poisoning, and should be continued until HbCO is <3% and the patient's presenting symptoms of CO poisoning have resolved. This is usually about 6 h.4
Because a higher inspired PO2 hastens the decrease in HbCO, it is reasonable to expect that hyperbaric oxygen should be of benefit for CO poisoning. Hyperbaric oxygen (HBO) at 2.5 atm decreases the half-life of HbCO to < 30 min.30 The study by Weaver et al31 is often cited in support of the use of HBO for CO poisoning. In that study, subjects with symptomatic acute CO poisoning were randomly assigned to 3 chamber sessions within a 24-h period, consisting of either 3 HBO treatments or one normobaric O2 treatment plus 2 sessions of exposure to normobaric room air. Subjects who received HBO had approximately half the rate of cognitive sequelae at 6 weeks, 6 months, and 12 months after treatment compared to those who were treated with normobaric O2. Based largely on this study, some authorities recommend that HBO should at least be considered in all cases of serious acute CO poisoning.4
A Cochrane review by Buckley et al32 concluded that existing randomized trials do not establish whether HBO therapy for patients with CO poisoning reduces the incidence of adverse neurologic outcomes. The authors also state that additional research is needed to better define the role, if any, of HBO in the treatment of patients with CO poisoning. On the other hand, Rose et al5 argue that the benefits of HBO are difficult to evaluate in a meta-analysis. In the Cochrane review, 3 studies used only 2 atm for HBO, which is not considered adequate, and only one of the studies evaluated neurocognitive outcomes > 1 month after poisoning,
The American College of Emergency Physicians published a clinical policy related to the management of adult patients presenting to the emergency department with acute CO poisoning.33 They recommend that HBO is a therapeutic option for CO-poisoned patients. However, its use is not mandated. They also state that no clinical variables, including HbCO, can identify a subgroup of CO-poisoned patients for whom HBO is most likely to provide benefit or cause harm. Thus, despite a well-done single-center study supporting benefit for HBO in patients with CO poisoning, this is an area of controversy.
Exhaled CO and Smoking Cessation
The HbCO level in smokers is generally in the 3–5% range.4 For each pack of cigarettes smoked per day, the HbCO increases about 2.5%,34 and HbCO > 20% has been reported with cigarette smoking.35 Exhaled CO is increased in cigarette smokers.15,18,36,37 Exposure to shisha smoke (via hookah) also causes elevated exhaled CO in active smokers and passive smokers.38,39 It follows that evaluation of HbCO might be useful to assess the success of smoking cessation efforts and the recidivism of those smokers who claim to have quit. Middleton and Morice40 suggest that an exhaled CO > 6 ppm strongly suggests that a patient is a smoker. Cunnington and Hormbrey41 found that subjects with exhaled CO > 6 ppm should be assessed for the risk of CO poisoning. However, even CO concentrations < 6 ppm do not exclude smoking within the last 24 h, and the range of breath CO values was large. It is interesting that the cut-points for classifying smokers versus nonsmokers varies among devices.17 Monitoring of exhaled CO adds value to smoking cessation programs and is included in the core competencies for evidence-based treatment of tobacco dependence (https://attud.org/pdf/Standards.pdf).
Inhaled CO to Evaluate Lung Function
The single-breath diffusing capacity for CO (DLCO) is commonly measured in the pulmonary function laboratory. This is a measure of the capacity of the lungs to exchange gas across the alveolar–capillary membrane. To measure CO diffusing capacity, the test gas typically includes 0.3% (3,000 ppm) CO.42 This high concentration is considered safe because it is a single breath of 10 s. HbCO can affect DLCO by an anemia effect and because CO partial pressure in the blood reduces the driving pressure for CO diffusion from the alveolus to the blood. However, adjustment is not required if HbCO is < 2% because reference equations already incorporate this. For HbCO > 2%, mathematical adjustments can be applied.42 For steady-state DLCO, CO at 0.1% (1,000 ppm) is breathed for 7 min.43 Most pulmonary function laboratories have abandoned the steady-state procedure in favor of the single-breath method.42
Diffusing capacity has been measured in mechanically ventilated patients with ARDS. Macnaughton and Evans44 used rebreathing measurements of DLCO in 15 subjects with ARDS. DLCO was significantly greater in survivors than in nonsurvivors. Di Marco and colleagues45 used a rebreathing method to measure DLCO in 16 subjects with ARDS. In subjects with ARDS, higher PEEP (15 cm H2O compared to 5 cm H2O) resulted in an increase in DLCO (4.4 ± 1.7 mL/mm Hg per min vs 3.6 ± 1.4 mL/mm Hg per min). In 8 subjects, there was >20% increase in DLCO with higher PEEP, and the PaO2/FIO2 increased only in these subjects. In a follow-up study, Pinto Da Costa et al46 evaluated DLCO in 16 subjects receiving pressure support ventilation. The baseline level of pressure support was 9.9 ± 2.1 cm H2O, with a DLCO of 4.4 ± 1.6 mL/mm Hg per min. The change in DLCO was not clinically important when pressure support was increased by 5 cm H2O.
Heme Oxygenase
Heme oxygenase (HO) is an important enzyme in human physiology. Heme oxygenase-2 (HO-2) is constitutively expressed (ie, continually; always on), whereas heme oxygenase-1 (HO-1) is strongly inducible (ie, off unless stimulated). HO catalyzes the first and rate-limiting step in heme degradation, yielding equimolar amounts of CO, iron, and biliverdin (Fig. 3), which is further converted to bilirubin by biliverdin reductase.47 The presence of HO explains the normal basal level of HbCO and bilirubin. Induction of HO-1 gene expression occurs in response to a wide variety of endogenous and exogenous stimuli,47 including hyperoxia,48 chemical or physical stimuli, foreign chemical compounds (xenobiotics), inflammation,49 surgical procedures, and anesthetics.50,51 The role of HO-1 is being investigated in the setting of acute lung injury,52 lung disease,53–55 pulmonary hypertension,56 sepsis,57–60 vascular disease,61,62 kidney disease,63 pancreatic disease,64 liver disease,65 gastrointestinal disease,66 solid organ transplantation,67 ischemia/reperfusion injury,68–70 and critical illness.71

The role of heme oxygenase in hemoglobin catabolism.
There are several published examples HO-1 expression in critically ill subjects. Hayashi and colleagues51 found that exhaled CO and arterial HbCO were increased on the day after patients underwent anesthesia and surgery, compared with preoperative values. In critically ill subjects, Scharte et al72 reported significantly higher exhaled CO (median 2.4 ppm) compared to healthy controls (median 1.55 ppm). CO production was also significantly higher in critically ill subjects (median 20 mL/min) compared to controls (median 13.5 mL/min). Fazekas et al73 found that subjects who survived an admission to the ICU had slightly higher minimum and marginally higher average HbCO levels when compared to nonsurvivors. Melley and colleagues74 found that both low minimum and high maximum levels of arterial HbCO were associated with increased ICU mortality. Thus, although the HO system is protective, excessive induction may be deleterious. This suggests that there may be an optimal range for HO-1 induction.
Due to HO activity, it might be expected that CO as well as bilirubin is increased in the presence of hemolysis. Lozar-Krivec and colleagues75 found that HbCO was higher in newborns with hemolysis than in those without hemolysis. The authors of several studies76–78 suggest that measurement of exhaled CO is a feasible approach to assessing hemolysis in jaundiced neonates. Exhaled CO might also be useful to monitor hemolysis in children with sickle cell disease.79
Physiologic Role for CO
The result of HO activity is related to its 3 reaction products: iron, bilirubin, and CO.80 The iron released triggers pathways involving the activation of the iron regulatory protein, and competing pathways involve its extracellular efflux and its potential participation in deleterious cellular reactions. The biliverdin and bilirubin produced contribute to cellular antioxidant balance prior to excretion. CO can confer modulatory effects on the regulation of vascular function, inflammation, apoptosis, and cell proliferation, and through stimulation of several signaling pathways.81 Similar to nitric oxide, CO can activate soluble guanylate cyclase to increase the production of cyclic guanosine monophosphate.82 In animal models of sepsis, CO exposure offers protection by inhibiting inflammatory responses and enhancing bacterial phagocytosis in leukocytes.60 Animal models have also shown that CO is protective against ventilator-induced lung injury.83–85
HO-1 induction seems important to mitochondrial biogenesis.86,87 Mitochondrial biogenesis involves coordination of expression, import, and assembly of mitochondrial proteins from nuclear and mitochondrial genomes and regulation of mitochondrial content and morphology.88 Carre et al89 evaluated mitochondrial biogenesis from muscle biopsies in 16 critically ill subjects and from 10 otherwise healthy subjects undergoing elective hip surgery. Subjects who survived responded early to critical illness with mitochondrial biogenesis (restorative) defense responses. In a murine model of sepsis, MacGarvey and colleagues90 administered CO at 250 ppm in air for 1 h, which resulted in mitochondrial biogenesis that was pro-survival and counter-inflammatory.
The cellular processes of apoptosis50,62,91 and autophagy60,92,93 might be potential therapeutic targets for CO. Apoptosis is a form of cell death in which a programmed sequence of events leads to the elimination of cells without releasing harmful substances into the surrounding area. It plays an essential role in the body by eliminating old cells, unnecessary cells, and unhealthy cells. Autophagy is a homeostatic process in cells to sequester cytoplasmic components in autophagosomes, where their contents are delivered for degradation and recycling. Autophagy acts as a survival mechanism under conditions of stress by regenerating metabolic precursors and clearing subcellular debris.
Inhaled CO as Therapy
Given the abundance of basic science supporting the therapeutic uses of CO, clinical trials are exploring this potential. Bathoorn et al94 explored the safety, feasibility, and anti-inflammatory potential of CO inhalation by stable COPD subjects. Subjects were treated with inhaled CO at 100–125 ppm for 2 h/d on 4 consecutive days. The median HbCO after the fourth inhalation of 100 ppm CO was 2.6%, with a highest individual value of 3.5%. After breathing 125 ppm, the median HbCO was 3.1%, with the highest individual value reaching 4.5%. CO inhalation led to trends in reduced sputum eosinophils and improved responsiveness to methacholine. As suggested by the authors, further studies are needed to confirm the safety and efficacy in inflammatory lung diseases.
A delivery system has been developed to administer CO in clinical trials (12th Man Technologies, Garden Grove, California).95 It is currently FDA-cleared only as an investigational device exemption for use in research protocols. This system injects CO into the gas flow (eg, ventilator system) similar to delivery of inhaled nitric oxide (Fig. 4).96 A 5,000-ppm CO source gas balanced with air is injected into the circuit in proportion to gas flow to provide a constant delivered CO concentration. The dose is confirmed by downstream measurement of the CO concentration using a calibrated sidestream electrochemical analyzer.

Carbon monoxide (CO) delivery system used in animal models and Phase 1 clinical trials. A: Front view. B: Schematic including ventilator, CO delivery system with injector module, and CO gas cylinder. C: Volumetric mixing data using the CO delivery system, a mechanical ventilator, and test lung. Flow-matching results in instantaneous flow of CO gas proportional to the inspiratory flow (Vent Flow) to deliver and maintain a precise and constant CO concentration. From Reference 95, with permission.
Fredenburgh et al95 used a clinically relevant baboon model of Streptococcus pneumoniae pneumonia to determine a safe and effective CO dosing regimen and to investigate the local and systemic effects of CO therapy on inflammation and acute lung injury. Inhaled CO was administered to 5 animals at 0, 6, 24, and/or 48 h after bacterial inoculation. The CO dose needed to achieve a HbCO target of 6–8% after 1 h of inhalation was determined, which was considered likely therapeutic while minimizing toxicity. The authors found that CO inhalation at 200 ppm for 60 min was well tolerated and achieved a HbCO of 6–8% with ambient CO ≤ 1 ppm. The HbCO measured at 20 min predicted the 60-min HbCO level by the CFK equation with high accuracy. Animals receiving inhaled CO and antibiotics had significantly less lung injury 8 d after inoculation compared with antibiotics alone. Inhaled CO was associated with activation of mitochondrial biogenesis in the lungs and with augmentation of renal antioxidative mechanisms. This suggests that CO might have both local and systemic protective effects. In the same model,97 lipid-specialized pro-resolving mediator profiles were partially restored with low-dose inhaled CO, which might shorten the time to pneumonia resolution.
A multi-center, prospective, randomized, placebo-controlled Phase 1 clinical trial of inhaled CO for the treatment of sepsis-induced ARDS is under way (NCT02425579). The results of this study will likely inform the future role of inhaled CO for the treatment of ARDS.
Summary
CO toxicity is an important cause of morbidity and mortality. The important role of heme oxygenase and endogenous CO production is an area of intensive investigation. Given the abundance of basic science supporting a therapeutic role for CO, clinical trials are exploring this potential.
Footnotes
- Correspondence: E-mail: dhess{at}aarc.org.
Dr Hess discloses relationships with Philips Respironics, Ventec Life Systems, Jones and Bartlett, McGraw-Hill, UpToDate, and the American Board of Internal Medicine.
Dr Hess presented a version of this paper at the 32nd New Horizons in Respiratory Care Symposium: Gases on October 16, 2016.
- Copyright © 2017 by Daedalus Enterprises
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.
- 7.
- 8.↵
- 9.↵
- 10.
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.
- 55.↵
- 56.↵
- 57.↵
- 58.
- 59.
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵









{kind=link}
{kind=link}
{kind=link}
{kind=link}