

Introduction
Non-Hodgkin lymphoma usually arises from lymphoid or nodal tissue, and can occasionally involve other sites. Extra-nodal marginal-zone B-cell lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma) is uncommon and constitutes around 8% of all non-Hodgkin lymphomas. Most MALT lymphomas are of gastric origin and lung involvement is rare. Primary pulmonary lymphoma is defined as clonal lymphoid proliferation, which can affect one or both lungs and/or bronchi, with no extrapulmonary involvement at diagnosis or within the subsequent 3 months.1,2 The majority (58–87%) of primary pulmonary lymphomas are low-grade MALT lymphomas; the rest are diffuse large-B-cell lymphomas and other high-grade lymphomas (11–19%).3,4
Patients with immunodeficiency are predisposed to non-Hodgkin lymphoma. In patients with human immunodeficiency virus (HIV), non-Hodgkin lymphoma is predominantly high-grade lymphoma, but a 14-fold relative risk of low-grade non-Hodgkin lymphoma has also been reported.1 Primary pulmonary lymphoma usually has an indolent course and can cause diagnostic dilemmas because of its nonspecific clinical and radiological presentation. We report a case of this rare MALT lymphoma that suggested a diagnosis of acquired immune deficiency syndrome (AIDS), and discuss the clinical, radiology, and pathology findings that can assist in the diagnosis.
Case Summary
A 51-year-old African American man presented with 3-weeks of dry cough and dyspnea, and a 3.6-kg weight loss. His medical history included COPD, hypertension, and treatment for latent tuberculosis. He was an ex-smoker, carpenter by occupation, and had been exposed to dust from the 2001 destruction of the World Trade Center. He was on inhaled steroids and bronchodilators. He had bilateral axillary lymphadenopathy and bilateral basal crepitations on lung auscultation, but the rest of the examination revealed no pertinent findings.
Complete blood count and comprehensive metabolic panel, including liver function tests, were within normal limits. HIV test via enzyme-linked immunosorbent assay (ELISA) was negative. Chest radiograph showed bilateral reticulonodular pattern, more prominent at the base of the lung (Fig. 1A). Computed tomogram showed left-lower-lobe pneumonia and left inter-lobar septal thickening, including thickened transverse fissure (Fig. 2). Pulmonary function testing revealed an obstructive pattern and low diffusing capacity for carbon monoxide. The differential diagnoses included community-acquired pneumonia, sarcoidosis, interstitial lung disease, lymphoma, and bronchioloalveolar carcinoma.
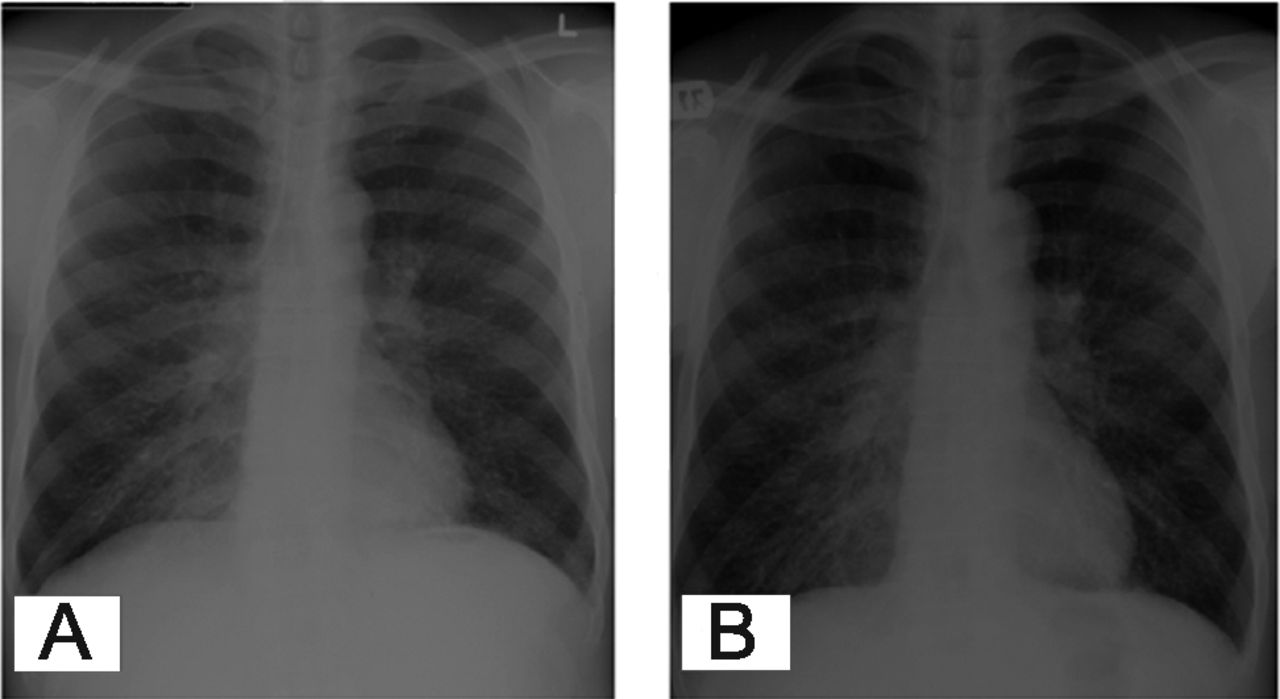
A: Radiograph shows bilateral reticulonodular pattern, more prominent at the lung base. B: Repeat radiograph shows worsening bilateral reticular changes prominent in the lung bases.
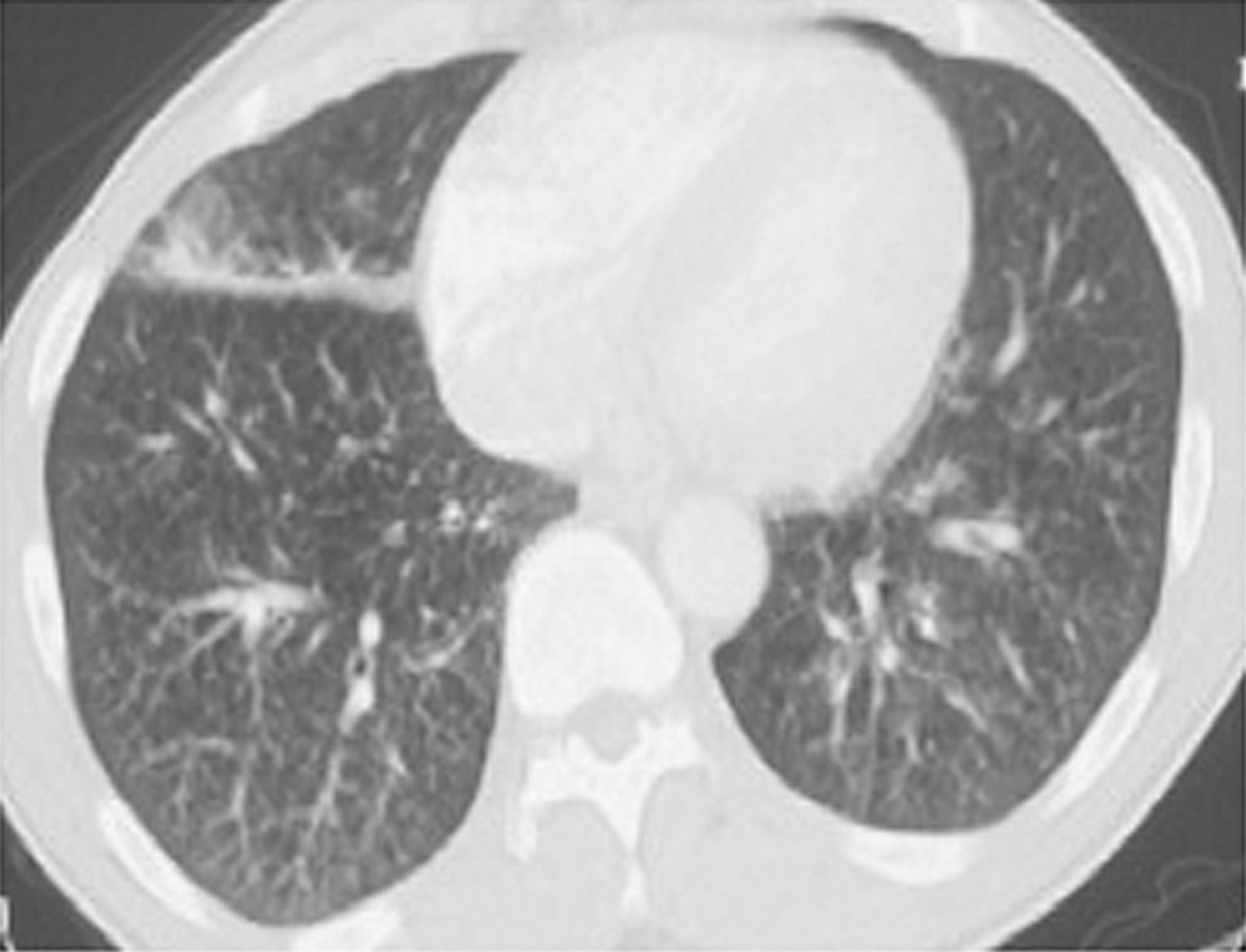
Computed tomogram shows septal thickening.
He was started on empiric antibiotics and discharged after reasonable clinical improvement, then was lost to follow-up. He was readmitted one year later with complaints of progressively decreased exercise tolerance and cough with whitish sputum. He was afebrile but tachycardic, tachypneic, and hypoxic (SpO2 92% on room air). He still had bilateral axillary lymphadenopathy and bilateral basilar crepitations. Complete blood count and comprehensive metabolic panel were within normal limits, except for elevated blood urea nitrogen (54 mg/dL). However, HIV (via ELISA test) was positive this time; his CD4 count was 341.
Chest radiograph showed bilateral reticular changes prominent in the lung bases (see Fig. 1B), compared to the previous radiograph. A subsequent computed tomogram showed increased subpleural and interseptal thickening. He was again started on empirical antibiotics (ceftriaxone and azithromycin) for suspected community-acquired pneumonia, along with sulfamethaxazole-trimethoprim and prednisone because of high clinical suspicion of Pneumocystis jirovecii pneumonia. Axillary lymph node biopsy showed follicular hyperplasia. Bronchial washings showed predominantly lymphocytes and were negative for Pneumocystis jirovecii pneumonia, acid-fast bacilli, and malignancy. Transbronchial lung biopsy showed chronic bronchial inflammation and preserved alveolar parenchyma. There was no granuloma or evidence of malignancy. Stains for acid-fast bacilli and fungi were negative.
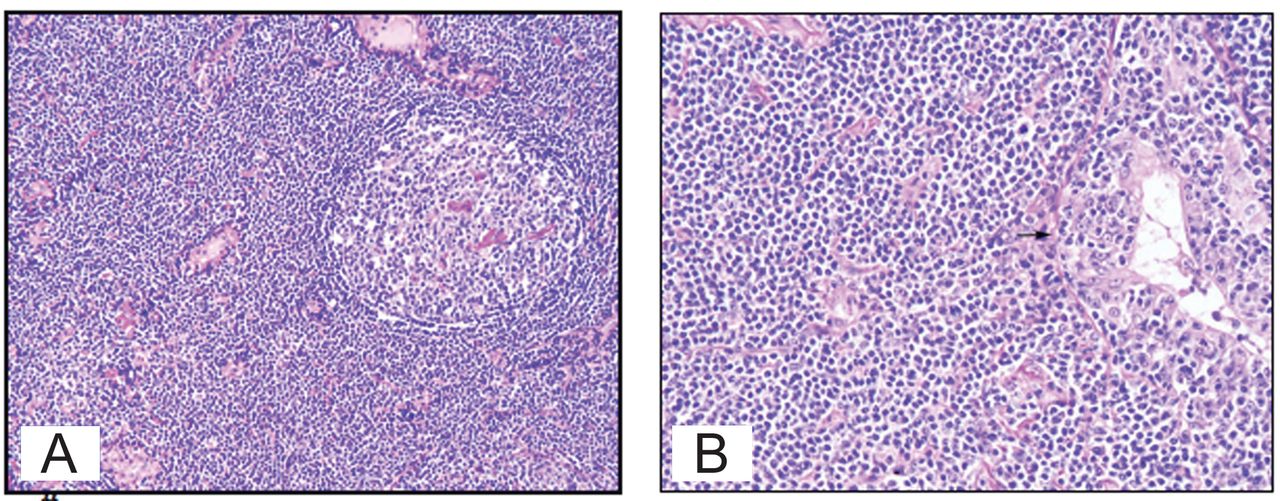
Video-assisted thoracoscopic lung biopsy showed moderate to marked interstitial chronic inflammation composed predominantly of small lymphocytes, plasma cells, and histocytes. There was also lymphoepithelial lesion eroding the bronchial mucosa, and focal lymphoid aggregates (Fig. 3), but no granulomas. Immunohistochemical studies showed prominent B-cell infiltrate positive for CD20 but negative for CD3 (T cell marker), cytokeratin and neuroendocrine markers, CD10, BCL6, BCL2, and CyclinD1. Polymerase chain reaction test showed immunoglobin (Ig) heavy-chain gene rearrangement, suggesting a neoplastic clonal proliferation of B cell. Fluorescence in-situ hybridization with Epstein-Barr virus early RNA probe was negative for Epstein-Barr virus.

A: A reactive B-cell follicle surrounded by neoplastic marginal-zone cells with an attenuated mantle cell cuff. B: Neoplastic marginal-zone cells infiltrating the bronchiolar epithelium and forming a lymphoepithelial lesion (arrow).
The diagnosis was low-grade pulmonary MALT lymphoma. Further workup failed to reveal any extra-pulmonary site of involvement. In view of the lack of literature supporting response to various therapies and poor adherence to therapy, we decided to closely monitor the patient. He was later started on anti-retroviral therapy, and the axillary lymphadenopathy resolved and he had overall symptom improvement. There was no change in the lung lesions on imaging at one year of follow-up.
Discussion
MALT lymphoma was first described by Issacson and Wright in 1983, in the gastrointestinal tract. In the latest World Health Organization classification,5 MALT lymphoma belongs to the marginal-zone lymphomas but is distinguished from the nodal and splenic forms by its different clinical behavior and cytogenetic characteristics. These tumors occur most commonly in the gastrointestinal tract, but they have been reported in other organs, including the salivary gland, head and neck, ocular adnexa, skin, thyroid, and breast.6 There are only a few case reports of pulmonary MALT lymphoma. Its pathogenesis is unclear, but chronic antigenic stimulation, as in autoimmune diseases, smoking, and infection, has been hypothesized as a trigger,7 which suggests that dysregulation of T cell function may be involved.
Similar mechanisms may be in play in HIV-positive patients who develop MALT lymphomas. In HIV-positive patients, some case reports and small series have reported MALT lymphomas at various sites, including the stomach, intestine, and thyroid gland, with secondary involvement of the lungs,8 but there are only rare reports of primary MALT of the lung in immunocompromised patients with AIDS.9
MALT lymphoma of the lung is most commonly diagnosed around age 60, possibly due to its slow growth and occult behavior, but there have been a few cases reported in the young. Our patient presented at age 51, which might be due to his altered immunity from HIV infection. Pulmonary MALT lymphoma patients are usually asymptomatic or have non-specific complaints, and the disease is often detected on routine chest radiograph. Symptomatic patients can present with cough, sputum, hemoptysis, chest pain, and/or moderate fever. Our patient had similar symptoms and physical signs, which contributed little to the diagnosis. The appearance of MALT lymphoma on plain chest radiograph is highly heterogeneous, but characterized by consolidation and well defined mass or reticulonodular pattern.2 Computed tomogram usually shows findings that are parenchymal, peripheral, predominantly in the lower zones, and bilateral/multiple.10 Pulmonary masses or consolidation or nodules, peribronchial or bronchovascular distribution, air bronchograms, dilated airways within the consolidated lung mistaken for cavitation, and pleural effusion have been described. Hilar or mediastinal lymphadenopathy is less common and suggests disseminated disease.9,10 Our patient had bilateral lower lobe involvement with consolidation and subpleural/interseptal thickening.
Bronchoalveolar lavage fluid (BALF) shows predominance of lymphocytes, and flow cytometric analysis helps differentiate it from reactive lymphocytic proliferation. Screening for monoclonal IgG in BALF supernatant (via immunoelectrophoresis) and restricted membranous or intracytoplasmic Ig light-chain expression (via slide immunohistochemistry) may be useful for diagnosis.11 The absence of a dominant B-cell clone in BALF could help dismiss invasive investigations of pulmonary lesions. In our patient, BALF was inconclusive. Histology of surgical biopsy is diagnostic in most cases. The biopsy can usually be via percutaneous transthoracic needle, but occasionally video-assisted thoracoscopic or open-lung biopsy is needed, as in our patient. The lymphoma cells are small to medium size, with irregular nuclear membranes, inconspicuous nucleoli, and moderate amounts of pale cytoplasm admixed with plasma cells, which predominantly infiltrate the marginal zone. In glandular tissues, the epithelium is often invaded by aggregates of lymphoma cells, resulting in lymphoepithelial lesion. These clonal cells express B-cell markers (CD20 and CD79a) with light-chain restriction, but are negative for T cell markers, CD5, CD10, CD23, and cyclin D1. The tumor cells usually show immunoglobulin heavy-chain rearrangement, via molecular studies.3,11
Localized MALT lymphoma has a good prognosis; the 5-year survival rate is > 80%. There is no consensus on treatment. There is no identified culprit antigen in the lung, contrary to the stomach (Helicobacter pylori), so antibiotics effective on low-grade localized gastric lymphoma are inappropriate. The treatment options are surgery, chemotherapy, and radiotherapy. Owing to a lack of comparative series, some authors have proposed watchful waiting, as we did in our patient.12 Occasionally, transformation to high-grade primary pulmonary lymphoma has been reported.
Surgical resection is preferred for localized tumors.13 Chemotherapy is generally used for patients with bilateral or extrapulmonary involvement, relapse, or progression. Combination regimens, such as cyclophosphamide, adriamycin, oncovin, and prednisone (CHOP), have not shown higher response rates than single-agent regimens with chloraminophene, cyclophosphamide, Azathioprine, or steroid.12,13 Radiotherapy is rarely used. The effect of anti-retroviral therapy on MALT lymphoma in patients with coexisting AIDS is not known.
With aggressive support and highly active anti-retroviral therapy for AIDS, HIV-positive patients are living longer, so it is likely that more cases of MALT and low-grade lymphoma will occur in these patients. Our case suggests that primary MALT lymphoma can be an early manifestation of AIDS or possibly that a latent primary pulmonary lymphoma can be unmasked by HIV infection. Thus, it is important to be sensitized to rare diagnoses such as these, because with time the biologic behavior of these lesions may become evident. The possibility of low-grade and MALT types of lymphoma should be considered when biopsy shows a dense small lymphocytic infiltrate in an HIV-positive patient. Immunologic and molecular analysis of tissue has a critical role in establishing the diagnosis.
Teaching Points
Patients with HIV infection are at higher risk for primary pulmonary lymphoproliferative diseases such as lymphocytic interstitial pneumonitis and primary pulmonary lymphoma, including MALT lymphoma.
In patients with HIV infection presenting with dyspnea and diffuse interstitial lung disease, lymphoproliferative diseases should be considered, after excluding opportunistic pulmonary infections.
BALF indicates lymphocytic alveolitis, specific tests for the diagnosis of lymphoproliferative disease should be pursued.
The diagnosis of extra-nodal MALT lymphoma is made based on morphologic and immunophenotypic analysis of lung biopsy, preferably surgical lung biopsy.
Lymphocytic interstitial pneumonitis (a polyclonal proliferation of lymphocytes in the lung parenchyma) is a common pulmonary complication of HIV infection and responds well to steroids.
Footnotes
- Correspondence: Vishal J Verma MD, Department of Pulmonary Medicine, The Brooklyn Hospital Center, 121 Dekalb Avenue, Brooklyn NY 11205. E-mail: vpv9002{at}nyp.org.
-
The authors have disclosed no conflicts of interest.
- Copyright © 2011 by Daedalus Enterprises Inc.





{kind=link}
{kind=link}
{kind=link}