

Abstract
Pulmonary hypertension is a rare disease in neonates, infants, and children, and is associated with substantial morbidity and mortality. An adequate understanding of the controlling pathophysiologic mechanisms is lacking. Moreover, a minority of research is focused specifically on neonatal and pediatric populations. Although therapeutic options have increased over the past several decades, they remain limited. In advanced pulmonary hypertension, progressive pulmonary vascular functional and structural changes ultimately cause increased pulmonary vascular impedance, right-ventricular failure, and death. Management includes the prevention and/or treatment of active pulmonary vasoconstriction, the support of right-ventricle function, treatment of the underlying disease (if possible), and the promotion of regressive remodeling of structural pulmonary vascular changes. Most currently available therapies augment or inhibit factors, or mediators of their downstream signaling cascades, that originate in the pulmonary vascular endothelium. These pathways include nitric-oxide/cyclic guanosine monophosphate (cGMP), prostacyclin, and endothelin-1. The ability to reverse advanced structural changes remains an as yet unattained goal. This paper reviews the epidemiology, pathophysiology, current treatments, and emerging therapies related to neonatal and pediatric pulmonary hypertension.
- pulmonary hypertension
- pediatric
- neonatal
- children
- infants
- endothelium
- pulmonary vascular disease
- rho kinase
- ROCK
Introduction
Pulmonary hypertension (PH) is defined as a mean pulmonary artery pressure (PAP) of ≥ 25 mm Hg at rest. This straightforward hemodynamic definition belies the diversity of conditions that can cause or contribute to the development of PH in neonates, infants, and children (Table 1). Unfortunately, an adequate understanding of the controlling pathophysiologic mechanisms for most forms of PH is lacking. Moreover, a minority of PH research is focused specifically on neonatal and pediatric populations, in whom outcomes depend on the etiology. Although therapeutic options have increased over the past several decades, they remain limited. In addition, clinically relevant pulmonary vascular dysfunction that precedes a substantial increase in PAP may be important in subsets of patients. Therefore, when reviewing recent advances in the management of PH, it is useful to place the discussion within the context of an expanding appreciation of the underlying pulmonary vascular biology, since perhaps the most promising advances are just emerging.
Clinical Classification of Pulmonary Hypertension
Clinical Classification and Etiology
As evident by the evolution of clinical classification schemes over the past 40 years, PH is not a single entity. The initial classification endorsed by the World Health Organization (WHO) in 1973 divided PH into only 2 categories: primary and secondary. The most recent classification, which followed the 4th world symposium on PH in 2008, divided PH into 5 groups and 27 subgroups (see Table 1).1 The rationale for the increasing subdivisions was to group patients who are similar in terms of clinical and pathophysiologic features and response to therapy.
The prevalence of PH in pediatric patients is not known precisely. A recent report from the United Kingdom Pulmonary Hypertension Service for Children described 64 children with idiopathic PH.2 From that cohort, the incidence of idiopathic PH was estimated to be 0.48 cases per million children per year, with a prevalence of 2.1 cases/million. Survival was reported to be 89%, 84%, and 75% at 1 year, 3 years, and 5 years, respectively. Factors predicting decreased survival included WHO functional class, and failure to thrive (based on height and weight z-score) at presentation. A French registry that captured 50 children, with a mean age of 8.9 years, between 2005 and 2006, estimated the prevalence of PH to be 3.7 cases/million.3 In that cohort, 60% had idiopathic PH, 24% had PH associated with congenital heart disease, and 10% had familial PH.3 An earlier report from the United Kingdom Pulmonary Hypertension Service for Children, from 2001 to 2006, described 216 children with PH.4 In that cohort, 28% of the patients had idiopathic PH, with a mean age at presentation of 7.37 years. Of those with associated PH (72%, mean age at presentation 7.9 years), 31% had Eisenmenger physiology, 30% had postoperative PH, 19% had PH associated with lung disease, 9% had PH associated with miscellaneous disorders (including human immunodeficiency virus [HIV]), bone marrow transplant, and metabolic disease, 6% had connective-tissue disease, and 5% had PH associated with complex un-operated or palliated congenital heart disease.4 Between 1999 and 2005, a Swiss registry enrolled patients with PH and identified 23 patients ages 0 to 18 years, with a mean age at diagnosis of 3 years.5 Of those patients, 52% had PH associated with congenital heart disease, 35% had idiopathic PH, and 13% had PH associated with pulmonary diseases. Similarly, 2 relatively large series (one European, the other 2 in the United States) that enrolled pediatric PH patients for drug-treatment trials described similar PH classification distributions.6,7
In the neonatal population, persistent pulmonary hypertension of the newborn (PPHN) warrants particular attention. The incidence of PPHN is approximately 2 per 1,000 live births.8 PPHN can be a primary disorder of the fetal pulmonary circulation, or secondary to pathologic processes that cause a maladaptive transition from the fetal to the neonatal circulation, such as meconium aspiration or surfactant deficiency, or diseases that result in abnormalities of lung development, such as congenital diaphragmatic hernia.9
In addition, there is an association between pulmonary arterial hypertension (PAH) and chronic hemolytic anemia, especially sickle-cell disease. The distinction between PAH and PH related to elevated pulmonary venous pressure, however, is less clear. Estimates of PAP based on echocardiographic measurements of tricuspid regurgitation jet velocity have indicated that approximately 30% of patients with sickle-cell disease have PH.10,11 However, catheter-based data indicated that almost half of the patients with PH had elevated pulmonary venous pressure or hyperkinetic states that resulted in elevated PAP with normal pulmonary vascular resistance (PVR).10 A recent study of 50 pediatric patients with sickle-cell disease used Doppler echocardiography combined with respiratory mass spectrometry to calculate pulmonary blood flow and stroke index, and to estimate PVR. Forty-four percent of the children with sickle-cell disease had a tricuspid jet velocity of ≥ 2.5 m/s (the definition of PH in the study), compared to 6% of age-matched normal controls. However, right-ventricular stroke volume was higher and PVR was lower in patients with sickle-cell disease and PH, compared to controls.12
Finally, it is important to recognize that patients may have substantial pulmonary vascular disease without having a resting PAP that meets the definition of PH.13 For example, patients with congenital cardiac defects resulting in either increased pulmonary blood flow or impaired pulmonary venous drainage are prone to episodes of acute reactive pulmonary vasoconstriction that can result in catastrophic cardiopulmonary collapse, particularly in the postoperative period after cardiopulmonary bypass.14,15
Pathophysiology
The pathophysiology of PH is multifactorial, complex, and incompletely understood. Various etiologies are associated with different particular mechanisms of disease, and a unifying construct has not been identified. However, several pathways common to a number of etiologies have been elucidated and have been leveraged in the development of novel and effective therapies.
Hemodynamics and Morphology
From a hemodynamic standpoint, the morbidity and mortality associated with PH relates to increased right-ventricular afterload. Over time, compensatory mechanisms fail, leading to right-heart failure and death. It is important to note that the tempo of this clinical sequence differs across etiologies and individual patients. For example, right-ventricular failure can develop rapidly in an infant following cardiac surgery (ie, postoperative pulmonary hypertensive crisis) or may progress over years in other patients (eg, Eisenmenger syndrome).
Although right-ventricular failure is a common final outcome in patients with PH, the pulmonary vascular changes are more diverse and relate to the specific etiology. This is important when considering available therapies, since therapies appropriate for one patient group may be deleterious for another. For example, inhaled nitric oxide (INO) may be effective for patients suffering from acute pulmonary arteriolar constriction (eg, PAH, PH owing to lung disease and/or hypoxia), but may be entirely ineffective or even harmful in patients with pulmonary veno-occlusive disease or left-heart failure.16–18
Among the various PH groups, the mechanisms that result in increased right-ventricular afterload are best understood in PAH. However, left-heart disease is a common cause of PH, at least in adults,19 in whom elevated PAP relates to the transmission of elevated left-atrial pressure. PVR may be normal. Although subsets of patients with left-heart disease develop PAH, the associated mechanisms are less well understood, and specific therapies for these patients have not been adequately studied.20–23 Likewise, the pulmonary vascular changes associated with pulmonary veno-occlusive disease, pulmonary capillary hemangiomatosis, and congenital cardiac defects associated with pulmonary venous obstruction are less well studied, but there are probably important differences from pathologies localized to the precapillary pulmonary vascular bed.24–26
In PAH, increased right-ventricular afterload relates to increased PVR and decreased compliance.27,28 Whereas historically hemodynamic assessments focused on measuring PAP and calculating PVR in PH patients, recent data have demonstrated value in measuring pulmonary vascular impedance, which combines resistance and compliance. In fact, some studies have found that impedance better predicts outcomes in PH patients than isolated measurements of PVR.29–31
Increased PVR and decreased compliance in PAH relate to several basic mechanisms: increased pulmonary vascular reactivity, sustained pulmonary vasoconstriction, vascular remodeling, and luminal obstruction due to in situ thrombosis and/or obstructive neointimal and plexiform lesions. Vascular remodeling involves changes of the medium and small pulmonary arteries, including medial wall hypertrophy, adventitial thickening, endothelial proliferation, and the abnormal extension of muscularization to normally non-muscular distal pulmonary arteries. These vascular changes tend to progress in a stepwise fashion, and in severe disease obliterate portions of the pulmonary circulation at the level of the distal precapillary resistance arterioles. It is recognized that this sequence represents a pathologic framework, but that substantial heterogeneity exists in terms of the precise pathology of PAH.32 Furthermore, the degree to which these changes are reversible remains unclear, but probably depends in part upon the etiology, and may be influenced by age.33 For example, in a seminal study, Rabinovitch and colleagues demonstrated that age at surgery, lung morphometric analysis, and the Heath-Edwards system grade predicted the reversibility of structural and functional pulmonary vascular changes secondary to congenital cardiac defects with increased pulmonary blood flow after surgical repair.34 In addition, it must be remembered that even early reversible pulmonary vascular disease can contribute to morbidity and mortality. An important study by Celermajer and colleagues, for example, demonstrated that children with increased pulmonary blood flow due to intracardiac shunting had impaired endothelium-dependent pulmonary vascular relaxation before their baseline PAP or PVR had significantly increased.13
PH associated with lung disease and/or hypoxemia is associated with vascular remodeling similar to PAH, but advanced obstructive plexiform lesions are less common. As expected, chronic thromboembolic PH is caused primarily by intraluminal obstruction.
Pulmonary Vascular Endothelium
Over the past 3 decades, an expansive body of literature has developed that has firmly established the central role of the pulmonary vascular endothelium in the pathophysiology of PH. In 1980, Furchgott and Zawadzki demonstrated for the first time the obligatory role of the vascular endothelium in systemic arterial smooth-muscle relaxation in response to acetylcholine.35 Subsequently it was shown that systemic and pulmonary vascular endothelial cells elaborate a number of factors that mediate both vascular smooth-muscle cell relaxation and constriction (Fig. 1).

Some endothelial-derived factors. These factors may increase and/or decrease smooth-muscle cell contraction. PGI2 = prostaglandin I2. PLA2 = phospholipase A2. AA = arachidonic acid. COX = cyclo-oxygenase. TXA2 = thromboxane A2. L-Arg = L-arginine. L-Cit = L-citrulline. NOS = nitric oxide synthase. ET-1 = endothelin-1. ECE = endothelin converting enzyme. ETA = endothelin A receptor. ETB = endothelin B receptor. NO = nitric oxide. sGC = soluble guanylate cyclase. GTP = guanosine-5′-triphosphate. cGMP = cyclic guanosine-3′-5′ monophosphate. AC = adenylate cyclase. ATP = adenosine-5′-triphosphate. cAMP = cyclic adenosine-3′-5′-monophosphate. PKA = protein kinase A. PKG = protein kinase G. PLC = phospholipase C. AMP = adenosine monophosphate. PDE = phosphodiesterase (type 3 and 5 shown). K+ = potassium channels. DAG = diacyl glycerol.
It is now accepted that increased pulmonary vasoconstriction in PH is mediated in large part by aberrant endothelial function, wherein endogenous vasodilators, such as nitric oxide and prostacyclin (PGI2), are decreased while endogenous vasoconstrictors, such as ET-1 and serotonin (5-HT), are increased (see Fig. 1).36–41 Indeed, the majority of approved therapies for PH target these endothelial-derived factors or their signaling pathways.
Nitric oxide is produced in the vascular endothelium by the enzyme endothelial nitric oxide synthase (eNOS), from the precursor L-arginine. Once formed, nitric oxide diffuses into the adjacent smooth-muscle cell and activates soluble guanylate cyclase (sGC), producing guanosine-3′-5′cyclic monophosphate (cGMP), which results in smooth-muscle cell relaxation through protein kinase G. cGMP is broken down by a family of phosphodiesterases (PDE). PDE5 is prominent in the pulmonary vasculature.
Arachidonic acid metabolism within vascular endothelial cells results in the production of PGI2 and thromboxane (TXA2). PGI2 activates adenylate cyclase, resulting in increased cyclic adenosine-3′-5′-monophosphate (cAMP) production, activation of protein kinase A, and consequent vasodilation, whereas TXA2 results in vasoconstriction via phospholipase C signaling. PGI2 also binds to platelet receptors, which inhibits their activation.
ET-1 is a 21 amino acid polypeptide that is also produced by vascular endothelial cells.42 The vasoactive properties of ET-1 are complex.43–47 However, its most striking property is its sustained hypertensive action. The hemodynamic effects of ET-1 are mediated by at least 2 distinct receptor populations: ETA and ETB.48,49 The ETA receptors are located on vascular smooth-muscle cells and mediate vasoconstriction, whereas the ETB receptors are located on endothelial and smooth-muscle cells and thus may mediate both vasodilation and vasoconstriction, respectively.
An important area of active research is focused on understanding the mechanisms responsible for endothelial injury or dysfunction in PH. Some important mechanisms include alterations in mechanical forces (such as increased pulmonary blood flow associated with congenital cardiac defects, or altered flow velocities that are associated with areas of luminal narrowing) that result in increased vascular-wall shear stress, hypoxia, oxidative stress, and inflammation.50–58 Additional factors that contribute to endothelial injury in some patients include, infection, such as HIV and schistosomiasis, and injury from drugs or toxins.59–61
Moreover, endothelial-derived factors, such as NO, PGI2, and ET-1, are integral to processes beyond the regulation of vascular smooth-muscle cell tone. Nitric oxide is a key regulator of vascular homeostasis and also has anti-thrombotic and anti-proliferative properties. Conversely, the mitogenic properties of ET-1 are well described. Indeed, endothelial injury or dysfunction probably contributes to alterations in inflammatory cascades, growth factors, and transcriptional factors that are increasingly recognized as key mediators of the vascular remodeling associated with PH.58 For example, alterations in platelet-derived growth factor (PDGF) and vascular endothelial growth factor (VEGF) contribute to abnormal smooth-muscle cell proliferation.62–65 Experimental models indicate important interactions between these factors. For example, in a rat model, VEGF inhibition impaired lung growth and resulted in PH: a result that can be reversed with nitric oxide.66 Finally, a disrupted endothelium is probably responsible, at least in part, for an increased propensity for intravascular thrombosis.
Pulmonary Vascular Smooth Muscle
Considerable efforts have been made to understand the processes responsible for the smooth-muscle cell hypertrophy and proliferation that accompany PH. Some known mechanisms include increased pericyte differentiation, smooth-muscle cell migration, endothelial cell transdifferentiation, smooth-muscle cell proliferation, smooth-muscle cell hypertrophy, and inflammation.67,68 However, increasing attention is now focused on also understanding potential alterations in the mechanisms that regulate smooth-muscle cell contraction. Myosin light chain (MLC) regulates smooth-muscle cell contraction. Myosin light chain kinase (MLCK) is responsible for the phosphorylation of MLC, leading to an increase in its activity, and myosin light chain phosphatase (MLCP) is responsible for the dephosphorylation of MLC. The activity of MLCK is regulated by Ca2+-calmodulin binding, so an increase in intracellular Ca2+ increases smooth-muscle cell contraction. Vasoconstrictors, such as ET-1 and thromboxane, increase inward Ca2+ flux by binding to G-protein coupled receptors.
In addition to Ca2+ concentration increase, contraction can be augmented by an increase in Ca2+ sensitization. RhoA is a small G-protein that activates downstream effectors, including the Rho kinases (ROCKs). One target of ROCK is myosin phosphatase target subunit (MYPT-1), which is the regulatory subunit of MLCP. Phosphorylation of MYPT-1 results in MLCP inhibition, decreased MLC dephosphorylation, and increased smooth-muscle cell contraction, at any given Ca2+ concentration. Beyond contraction, ROCKs appear to be important in smooth-muscle cell differentiation, proliferation, migration, and apoptosis.69 In fact, ROCKs have been shown to participate in vascular endothelial processes as well.69 Interest in Rho kinase inhibitors as a treatment for PH is increasing as positive results have been obtained from multiple animal studies.70–75
Genetics
It is now clear that underlying genetic abnormalities participate in the development of PH in some patients. Most prominently, mutations in bone morphogenetic protein receptor 2 (BMPR2) are associated with PAH in subsets of patients with and without a family history of disease.76–81 On the other hand, a minority (approximately 20%) of patients with BMPR2 mutations develops PAH, suggesting that the mutation alone is not sufficient to cause disease in most cases.80,82 The bone morphogenetic proteins are members of the transforming growth factor-β superfamily.77 The BMPR2 gene is located on chromosome 2q33, and multiple mutations have been identified. Inheritance patterns indicate autosomal dominant disease with incomplete penetrance and genetic anticipation. Estimates of BMPR2 mutation in pediatric PH vary, but a recent study that included 78 children with idiopathic PH identified 8 patients with BMPR2 mutations, and indicated that in adults and children BMPR2 mutations were associated with increased disease severity.83,84 Less common genetic associations include mutations in the activin-like kinase type-1 receptor (ALK-1) and endoglin genes.85,86
Management Strategies and Therapeutic Options
The goals of therapy depend on the clinical situation, but basic elements of PH management can be distinguished and applied as appropriate. These include the prevention and/or treatment of active pulmonary vasoconstriction, the support of right-ventricle function, treatment of the underlying disease (if possible), and the promotion of regressive remodeling of structural pulmonary vascular changes. Of course these aims are interrelated, but certain therapies may target one aim more than another in given patients. Table 2 summarizes the therapies described below.
Therapeutic Approach to Pediatric Pulmonary Hypertension
Prevention and/or Treatment of Active Pulmonary Vasoconstriction
Increased pulmonary vascular reactivity is an early feature of PH, which manifests clinically as augmented pulmonary vasoconstriction in response to such stimuli as hypoxia, acidosis, catecholamine-mediated α1-adrenergic stimulation associated with pain and/or agitation, and increases in intrathoracic pressure.87–89
In critical care settings, acute life-threatening pulmonary hypertensive crises are often first treated with pain control, sedation, oxygenation, and alkalinization. Studies in newborn lambs found dose-dependent pulmonary vasodilation in response to increasing pH, from 7.30 to 7.60, and dose-dependent response to increasing FIO2, from 0.21 to 0.50.90 Indeed, recently published clinical practice guidelines for the hemodynamic support of pediatric and neonatal septic shock specifically address the risk of elevated PAP/PVR and right-heart failure in neonates with sepsis, and the potential need for metabolic and respiratory alkalinization as a part of the initial resuscitation strategy.91 However, chronically, such therapies can be detrimental. For example, aggressive hyperventilation and oxygenation can promote the development of secondary lung injury in neonates with PPHN.92,93 As such, gentle ventilation, with mild permissive hypercapnia, lower FIO2, and INO with or without high-frequency ventilation, has become accepted practice for neonates with severe PPHN.93–95
Acute selective pulmonary vascular relaxation may be the primary therapeutic goal in some types of PH, such as severe PPHN or postoperative PH. On the other hand, in many other forms of PH, particularly chronic advanced PH, active vasoconstriction is not a prominent pathologic feature. For example, studies of pediatric patients with chronic PH have found a wide range of responsiveness to acute vasodilator testing, ranging from 7% to 40%.4,96 The proportion of adults with chronic PH who respond to acute vasodilators is probably even less.97
Endothelial-Based Therapies.
The most widely used PH therapies work by altering one of 3 endothelial signaling cascades: nitric oxide-cGMP, PGI2, or ET-1. Figure 2 is a simplified depiction of the various sites of action of the therapies.

Sites of action of some endothelial and smooth-muscle cell based therapies. Arrows indicate activation. T symbols indicate inhibition. Terms are as in Figure 1.
Nitric Oxide-cGMP Cascade: Inhaled Nitric Oxide.
INO is the best-studied and most widely used agent for acute selective pulmonary vasodilation. When delivered via inhalation, nitric oxide diffuses across the alveolus into the smooth muscle of the accompanying capillary, resulting in relaxation. Nitric oxide then diffuses into the blood vessel, where it is rapidly inactivated by its interaction with hemoglobin. In this way, the effects of INO are relatively confined to the pulmonary circulation and to ventilated areas of the lung, thus optimizing ventilation-perfusion matching. In large trials, INO decreased the need for extracorporeal life support in neonates with PPHN, and those data led to INO's FDA approval.98–100 Now INO is used to treat many other forms of PH, and for diagnosis. For example, several studies have investigated INO in pediatric patients undergoing cardiac surgery,15,101–104 and found INO effective in lowering PAP and PVR in the postoperative period, but the data were less clear about the impact on outcome.105 Likewise, investigators have examined the utility of INO in the particular situations of bidirectional cavopulmonary connections and after Fontan procedure.106–108 In those patients, INO decreased central venous pressure and transpulmonary gradient, and increased oxygen saturation. In addition, the pulmonary vascular response to INO has been studied as a part of the assessment for operability in patients with PH associated with congenital heart disease.109–112 Those studies found that the combination of 100% oxygen and INO (at 80 ppm) produced maximal pulmonary vasodilation and was more predictive than either treatment alone for postoperative outcome.109–112
Nitric Oxide-cGMP Cascade: PDE5 Inhibition.
Sildenafil is a PDE5 inhibitor, so its mechanism of action is to augment nitric oxide-cGMP signaling by inhibiting the degradation of cGMP. Increased cGMP results in pulmonary vascular relaxation. Sildenafil has both pulmonary and systemic effects, but the importance of this is not yet entirely clear. For example, systemic peripheral vascular relaxation could improve cardiac output, but non-specific pulmonary vascular relaxation could antagonize hypoxic pulmonary vasoconstriction, worsening hypoxia. In addition, the effects of PDE5 inhibition may not be restricted to the vasculature. For example, a recent study found that PDE5 was up-regulated in the hypertrophied right ventricle and that PDE5 inhibition improved contractility.113 Randomized placebo-controlled clinical trials, with one that included some pediatric patients, found oral sildenafil effective for the treatment of chronic PAH, in terms of improvements in functional outcomes (eg, 6-minute walk), time to clinical worsening, and hemodynamics.114–117 A study that enrolled 14 children with PH in an open-label clinical trail of sildenafil found similar results.118
Despite limited data, the use of sildenafil in infants and children with PH after cardiac surgery is increasing. Three small studies found that enteral sildenafil facilitated weaning from INO in pediatric patients with congenital heart disease undergoing therapy for postoperative PH.119–121 The 2 studies of intravenous sildenafil in pediatric patients after cardiac surgery122,123 found both that it decreased PAP and PVR, either to a greater extent than INO or synergistically, but that its use was associated with increased intrapulmonary shunt and decreased systemic arterial pressure.
Sildenafil has also been studied in a placebo-controlled, randomized trial in neonates with severe PPHN.124 Thirteen neonates with an oxygenation index of > 25 received either placebo or enteral sildenafil. The oxygenation index decreased by 30 hours in all 7 sildenafil patients, and 6 of those 7 neonates survived. There was no systemic hypotension in either group. In the placebo group, only 1 of the 6 patients survived. Likewise, in an open-label, dose-escalation trial that included 36 newborns with PPHN and an oxygenation index > 15 (most of whom were receiving INO), a continuous intravenous infusion of sildenafil for 2–7 days improved oxygenation index by 4 hours at higher doses.125
Newer PDE5 inhibitors (ie, tadalafil and vardenafil) offer longer half-lives as the primary potential advantage. In a recent randomized, placebo-controlled, open label study that included patients between 12 and 65 years of age with PAH, vardenafil improved exercise capacity and cardiac index, and decreased PAP and PVR.126 However, to date no studies have been published on younger pediatric PH patients.
Nitric Oxide-cGMP Cascade: Arginine and Citrulline Supplementation.
The administration of additional substrate for NOS is another approach to augmenting the nitric oxide-cGMP cascade. A study that included 19 adult patients with PAH, found that oral L-arginine supplementation improved exercise capacity and hemodynamics.127 These results confirmed an earlier study of 10 patients with PAH, which found that a short-term intravenous infusion of L-arginine produced a short-term decrease in PAP and PVR.128
L-arginine and citrulline levels are decreased in neonates with PPHN, in adults and children with PH related to sickle-cell disease, and in infants and children after cardiac surgery.129–132 In a small study, 5 neonates with PPHN received a single dose of intravenous L-arginine, which decreased the oxygenation index and increased oxygen saturation.133 In another study, 10 patients (age range 13–63 years) with sickle-cell disease were treated with oral L-arginine for 5 days and had a 15% decrease in estimated PAP.134
A study that included 20 infants with PH related to cardiac defects with increased pulmonary blood flow examined the effects of an infusion of intravenous L-arginine on hemodynamic variables.135 Ten patients were studied preoperatively and another 10 were studied 2 hours after separating from cardiopulmonary bypass in the postoperative period. PVR decreased in both groups in response to oxygen. However, a further decrease in response to L-arginine was seen only in the postoperative group.
Finally, a randomized, double-blind, placebo-controlled trial evaluated the effects of perioperative oral citrulline supplementation in 40 infants and children undergoing cardiac surgery.136 Citrulline level above 37 μmol/L appeared to protect against the development of postoperative PH.
Nitric Oxide-cGMP Cascade: Direct Soluble Guanylate Cyclase Activators.
Direct activation of sGC is a novel approach that is under investigation for the treatment of PH. A recent study found that sGC expression was increased in explanted lungs of patients with PAH.137 In animal models of PH, sGC expression was similarly up-regulated, and direct sGC activation improved hemodynamics and vascular remodeling.137–139 A proof-of-concept study was conducted that included 19 adult patients with PH (either PAH or chronic thromboembolic PH) who received the new drug riociguat (BAY 63–251).140,141 That study suggested that riociguat was safe and found that pulmonary hemodynamics and cardiac output improved in a dose-dependent fashion.140 A subsequent phase II study that included 75 adult patients with PAH or chronic thromboembolic PH found that riociguat improved exercise capacity and decreased PVR.142
Clinical studies in pediatric PH patients are not yet available. However, because these novel agents can activate sGC independent of NO, they are promising, since a decrease in bioavailable NO appears to be central to the pathophysiology of some forms of PH. In addition, NO binding to sGC is impaired when the heme group on sGC is oxidized.143 Interestingly, these agents can function when sGC is in the oxidized state,144 which may be important, given growing evidence of the role of oxidative stress in many forms of PH.
Prostanoids.
Higenbottam and colleagues first described the long-term use of intravenous PGI2 for the treatment of PH almost 30 years ago.145 Despite the many recent advances in therapy, intravenous PGI2, epoprostenol, remains the best-proven and most effective therapy for chronic PH.146–148 Yung and colleagues recently described a group of 35 children with idiopathic PAH treated with epoprostenol.149 In that cohort, survival at 1 year, 5 years, and 10 years was 94%, 81%, and 61%, respectively. This is particularly important given that the median survival in children with primary PH has been reported to be as low as 10 months without treatment.150 Interestingly, studies indicate that patients without an initial vasodilating response to PGI2 often gain important long-term benefit, which suggests that effects beyond vasodilation, such as anti-platelet effects, cAMP-mediated inhibition of smooth-muscle cell growth, or other unknown mechanisms may be responsible for the treatment effect.96 Complications associated with long-term epoprostenol are well known and include thrombosis and infection secondary to the required indwelling central venous catheter, the need for dose escalation, and life-threatening rebound PH after abrupt discontinuation of the infusion.
In addition to epoprostenol, treprostinil is another PGI2 analog that was recently approved for intravenous administration.151 Data are not available to compare the efficacy of intravenous epoprostenol to treprostinil. However, Ivy and colleagues reported the successful transition of 13 children from intravenous epoprostenol to treprostinil without changes in exercise capacity or intolerable adverse effects.152
Less is known about the use of intravenous prostanoids in pediatric patients with PH secondary to other conditions. Rosenzweig and colleagues reported on the use of long-term epoprostenol in 20 children with PH secondary to congenital cardiac defects. PAP and PVR decreased, and cardiac output and exercise capacity increased after one year of therapy.153
Given the success of intravenous epoprostenol, recent efforts have focused on developing additional agents and delivery approaches, in large part to address the complications and limitations associated with long-term intravenous infusions. To achieve selective pulmonary vascular relaxation, various investigations have focused on delivering prostanoids via inhalation.154–156 In a report on 4 newborns with PPHN refractory to INO, Kelly and colleagues described improved oxygenation with inhaled epoprostenol.155 Iloprost is a PGI2 analog that is FDA approved for administration via nebulization. Ivy and colleagues studied iloprost in 22 children with PH.157 They found that inhaled iloprost decreased PAP to a degree equivalent to INO with oxygen. A minority of patients (35%) demonstrated improved functional class, but up to 10% experienced acute bronchoconstriction. Likewise, Rimensberger and colleagues administered inhaled iloprost and INO, alone and in combination, to 15 children with PH secondary to congenital cardiac defects.158 Both agents decreased the PVR/SVR ratio to a similar degree, and there was no added benefit from a combination of the treatments. Furthermore, in an interesting study by Limsuwan and colleagues, in Thailand, which has less access to INO, inhaled iloprost decreased mean PAP and increased systemic saturation without decreasing systemic blood pressure in 8 children suffering from acute PAP increases after repair of congenital heart disease.159 In addition, treprostinil, which was initially approved by the FDA for subcutaneous delivery, was recently approved for delivery via inhalation. Several studies have indicated that inhaled treprostinil improves exercise capacity and decreases PVR and PAP in adults with chronic PH.160–162 More recently, a randomized controlled trial that included 212 adult patients with PH who were being treated with either bosentan or sildenafil, found that the addition of inhaled treprostinil improved exercise capacity, quality of life, and N-terminal pro-brain natriuretic peptide (NT-proBNP) levels, without changing pulmonary hemodynamics.163
Other dosing strategies for prostanoids include subcutaneous and oral administration. A number of studies, including randomized, placebo-controlled clinical trials have demonstrated the efficacy of subcutaneous treprostinil in improving exercise capacity in adults with PH.164–169 In children, a primary impediment to the use of subcutaneous treprostinil relates to pain at the injection site. Nonetheless, subcutaneous treprostinil has been used successfully in pediatric patients.170,171 Finally, oral prostanoid therapies are being developed. These include beraprost, which is a PGI2 analog, and an oral formulation of treprostinil. Results with beraprost have been mixed: several clinical trials have shown initial improvement in exercise capacity that may not be sustained over the long term.172–174
Endothelin-1.
Unlike augmentation of the NO/cGMP and prostanoid cascades, inhibition of endothelin-1 (ET-1) signaling does not reliably cause acute pulmonary vascular relaxation. However, in a small study that included 7 infants who had undergone surgical repair of left-to-right intracardiac shunt, Schulze-Neick and colleagues demonstrated that an intravenous infusion of a selective ETA-receptor antagonist resulted in an acute decrease in PVR.175 Notably, the addition of INO had no effect, and the decrease in PVR correlated with left-atrial ET-1 level. But, currently, intravenous ET-receptor antagonists are largely restricted to experimental settings.
Bosentan is an oral dual ET-receptor antagonist. Several double-blind, placebo-controlled, randomized clinical trials have confirmed the efficacy of bosentan in adult patients with PAH.176–178 These studies found, variously, that bosentan improved pulmonary hemodynamics, exercise capacity, and cardiac output while lengthening the time to clinical worsening.
In a study of 40 children with idiopathic or associated PAH, Maiya and colleagues found that bosentan improved exercise capacity and weight-gain in children with associated PAH, but in only 40% of children with idiopathic PAH.179 Rosenzweig and colleagues performed a retrospective study that included 86 children with idiopathic or associated PAH,7 and bosentan was associated with improved WHO functional class in 46% of the patients.
ETA receptors are located on vascular smooth-muscle cells and mediate constriction, but ETB receptors may mediate relaxation or constriction, depending on whether they are located on endothelial or smooth-muscle cells, respectively. In addition, ETB receptors are involved in the clearance of ET-1. Therefore, there is rationale for ETA-specific receptor antagonism for the treatment of PH. However, the degree to which ETB receptors mediate constriction or relaxation relates to the density of receptors on the smooth-muscle or endothelial cells, and this is a dynamic process. For example, in a lamb model of increased pulmonary blood flow, created by a systemic to pulmonary shunt, ETB localization to smooth-muscle cells increased by 8 weeks of age, with an associated increase in ETB-receptor-mediated pulmonary vasoconstriction.180 Moreover, Bauer and colleagues found that ETB-receptors were selectively up-regulated and appeared to be localized to the hypertrophied medial layer of pulmonary arteries in 14 patients undergoing pulmonary thromboendarterectomy for chronic thromboembolic PH.181
Two oral ETA-receptor antagonists have been studied for the treatment of PH: sitaxsentan and ambrisentan. Several studies demonstrated that sitaxsentan was efficacious for the treatment of idiopathic and associated PAH.182–185 Unfortunately, sitaxsentan has been taken off of the market, due to concern about hepatic toxicity in some patients. In fact, potential hepatic toxicity is associated with both bosentan and sitaxsentan, which are sulfonamide-based agents and metabolized by cytochrome P450 enzymes.186,187 Ambrisentan is a newer oral selective ETA-receptor antagonist that is metabolized by hepatic glucuronidation, with less involvement of the P450 enzyme pathway.188 Moreover, unlike bosentan and sitaxsentan, ambrisentan does not induce or inhibit the P450 enzymes.188 In fact, the successful use of ambrisentan has been reported in patients who discontinued bosentan and sitaxsentan due to elevated hepatic transaminase levels.189 Data are not yet available on the efficacy of ambrisentan for children under 12 years of age with PH. However, the results of 2 double-blind, placebo-controlled, randomized trials that were run concurrently were recently reported.182 The studies included 202 and 192 adult patients with PAH and found that ambrisentan therapy improved exercise capacity, cardiac index, time to clinical worsening, and Borg dyspnea score, and decreased PAP and B-type natriuretic peptide level.
Combination Therapy.
The rationale for combination therapy for PH stems from the discovery of multiple pathways that contribute to the pathophysiology of PH, the need for dose escalation with several therapies coupled with adverse effects at higher doses, and the relatively modest treatment effects of monotherapy without an acceptable impact on mortality.
A number of randomized, placebo-controlled, clinical trials have been conducted on various therapy combinations. The Bosentan Randomized Trial of Endothelin Antagonist Therapy for PAH (BREATHE-2) study included 33 adult patients with PAH, and compared epoprostenol with bosentan to epoprostenol alone.190 That study found no benefit, and a trend toward more adverse effects with combination therapy. The STEP-1 trial included 67 adult patients with PAH, and compared bosentan and iloprost to bosentan alone.191 There was an improvement in functional class and a delay in time to clinical worsening, but those results were not replicated by the Combination Therapy of Bosentan and Aerosolized Iloprost in Idiopathic Pulmonary Arterial Hypertension (COMBI) trial that compared those same treatments in 40 adult PAH patients.191,192 Channick and colleagues investigated the effects of adding inhaled treprostinil to bosentan therapy in 12 patients with PAH.160 That combination improved pulmonary hemodynamics, exercise capacity, and functional class.
The combination of sildenafil and prostanoids has also been investigated. The Pulmonary Arterial Hypertension Combination Study of Epoprostenol and Sildenafil (PACES) trial compared epoprostenol plus sildenafil to epoprostenol alone in 267 adult patients with PAH.193 The combination had favorable results, including better exercise capacity, pulmonary hemodynamics, quality of life, and a delay in time to clinical worsening. Likewise, Ghofrani and colleagues investigated the impact of sildenafil in 73 adult patients with PAH who were receiving inhaled iloprost.194 The addition of sildenafil improved exercise capacity and pulmonary hemodynamics. In a study meant to capture patients early in the disease course, patients with PAH at a WHO functional class of II were treated with bosentan. A subset of patients was receiving background sildenafil, and they had better pulmonary hemodynamics and time to clinical worsening.195 Several other trials have compared various combination therapies, and the preliminary results suggest benefits. Unfortunately, similar randomized trials in pediatric patients are not available, although many pediatric patients receive combination therapy.
Novel Endothelial-Based Therapies.
eNOS Coupling.
A decrease in bioavailable nitric oxide is an important component of endothelial dysfunction in PH. Several factors can decrease the bioavailability of nitric oxide, including decreased production by eNOS. When substrate or cofactors are limited, or due to oxidative stress, eNOS can become uncoupled, and then it produces superoxide anion instead of nitric oxide,54,196 which increases oxidative stress. Cicletanine improves eNOS coupling, thereby increasing bioavailable nitric oxide.197,198 At this point, the use of cicletanine in PH is limited to a case report, which described a positive response in an adult.199
Superoxide Dismutase.
Oxidant stress is well known to contribute to the endothelial dysfunction associated with vascular disease, including PH.54,200 Superoxide dismutase, the enzyme responsible for the clearance of superoxide anion, has been used clinically, including in premature infants.201–203 Steinhorn and colleagues found that recombinant human superoxide dismutase decreased PVR and was synergistic with INO in a fetal lamb model of PPHN.204
Cell-Based Therapy.
In 1997, endothelial progenitor cells (EPCs) were first isolated from human blood, and autologous EPCs were shown to incorporate into vessels during angiogenesis in animal models.205 Subsequently, therapeutic administration of EPCs was shown to ameliorate PH in animal models, with improved efficacy when EPCs were transduced with eNOS.206–208 In addition, several studies have found decreased and dysfunctional EPCs in patients with PAH.209,210 Wang and colleagues performed a prospective randomized trial to assess the safety and efficacy of adding autologous EPCs infusions to conventional therapy in 31 patients with idiopathic PH, with encouraging results.211 Specifically, patients demonstrated improved hemodynamics and exercise capacity, and there were no adverse events during the study period. More recently, that research group performed a pilot study in which 13 children with idiopathic PAH received an infusion of autologous EPCs.212 Twelve weeks after treatment the children had a modest PAP decrease, a 19% PVR decrease, increased cardiac output, improved exercise capacity, and an improvement in functional class.
An intriguing recent study by Smadja and colleagues quantified endothelial colony-forming cells (ECFCs, also called “late EPCs”) in 79 children with PAH. ECFCs were increased in the 8 children treated with treprostinil, compared to the rest of the cohort treated with oral ET-1 receptor antagonists and/or PDE5 inhibitors.213 In addition, ECFC cultured from these patients demonstrated a hyperproliferative phenotype with increased angiogenic potential.
Novel Vasodilators.
Vasoactive intestinal peptide (VIP) and adrenomedullin are 2 peptides with potent vasodilatory properties. A knock-out-mouse model demonstrated that deletion of the VIP gene led to moderate PAH under normoxic conditions.214 Moreover, Petkov and colleagues found that VIP was decreased in the serum and lung tissue of patients with PAH,215 and inhaled VIP decreased PAP and PVR, and increased cardiac output in 8 patients with PAH. Of note, these patients did not demonstrate a similar response to INO. Furthermore, after 3 months of daily treatment with inhaled VIP, these patients had improved pulmonary hemodynamics, cardiac output, exercise capacity, and Borg dyspnea score. Continued benefit was observed at 6 months, with further improvements in exercise capacity and decreases in the Borg dyspnea index.
Kakishita and colleagues found that adrenomedullin, a peptide with vasodilatory, angiogenic, anti-inflammatory, and anti-proliferative properties, was increased in adult patients with severe PH, and that adrenomedullin level correlated with hemodynamics including right-atrial pressure, stroke volume, PVR, and PAP.216,217 Nagaya and colleagues examined the acute effects of inhaled adrenomedullin in 11 adult patients with idiopathic PH,218 and adrenomedullin was associated with an acute decrease in PAP and PVR and increase in peak oxygen consumption during exercise.
Anticoagulation.
In patients with PH, in situ thrombosis is thought to contribute to pulmonary vascular remodeling and PVR elevation. Studies suggest that warfarin improves survival in patients with PAH, perhaps particularly in those with more advanced disease (ie, those unresponsive to acute vasodilator testing).219–221 Aspirin has no proven benefit, and other agents have not been adequately studied.
Smooth-Muscle-Based Therapy.
Calcium-channel blockers have long been used as a part of the evaluation and management of PAH, although their use is decreasing. Rich and colleagues demonstrated almost 20 years ago that a subset of patients with idiopathic PH had an acute vasodilatory response to calcium-channel blockers, and that in those patients high-dose calcium-channel blockers improved survival.221 More recent data, however, suggested that relatively few patients with idiopathic PH gain important long-term benefit.97 In addition, oxygen and INO have supplanted calcium-channel blockers for acute reactivity testing. Most recently, Montani and colleagues described 663 consecutive patients with associated PAH.222 A minority were responsive to acute vasodilator testing, and even fewer patients demonstrated a long-term response to calcium-channel blockers. In addition, there was variability between conditions. For example, there was a long-term benefit in 9.4% of acute responders with PAH associated with anorexigen, but less than 1% with PAH associated with portal hypertension or connective tissue disease. In fact, all 5 of the patients with pulmonary veno-occlusive disease or pulmonary capillary hemangiomatosis who responded to acute vasodilator testing deteriorated with calcium-channel blockers.
Rho-kinase inhibition is a novel therapeutic approach that targets the pulmonary vascular smooth muscle. Alterations in rho-kinase signaling have been demonstrated in several animal models of PH. In addition, Guilluy and colleagues demonstrated increased RhoA and Rho kinase activities in the lungs, platelets, and pulmonary artery smooth-muscle cells harvested from patients with idiopathic PH.223 As opposed to endothelial-based therapies, therapy aimed at altering Rho kinase activity has the potential advantage of directly affecting the contractile apparatus of the smooth-muscle cell (Fig. 3). For example, in animal models of severe PH, Rho kinase inhibition resulted in vascular relaxation in animals unresponsive to nitric oxide or PGI2 analogs.224,225
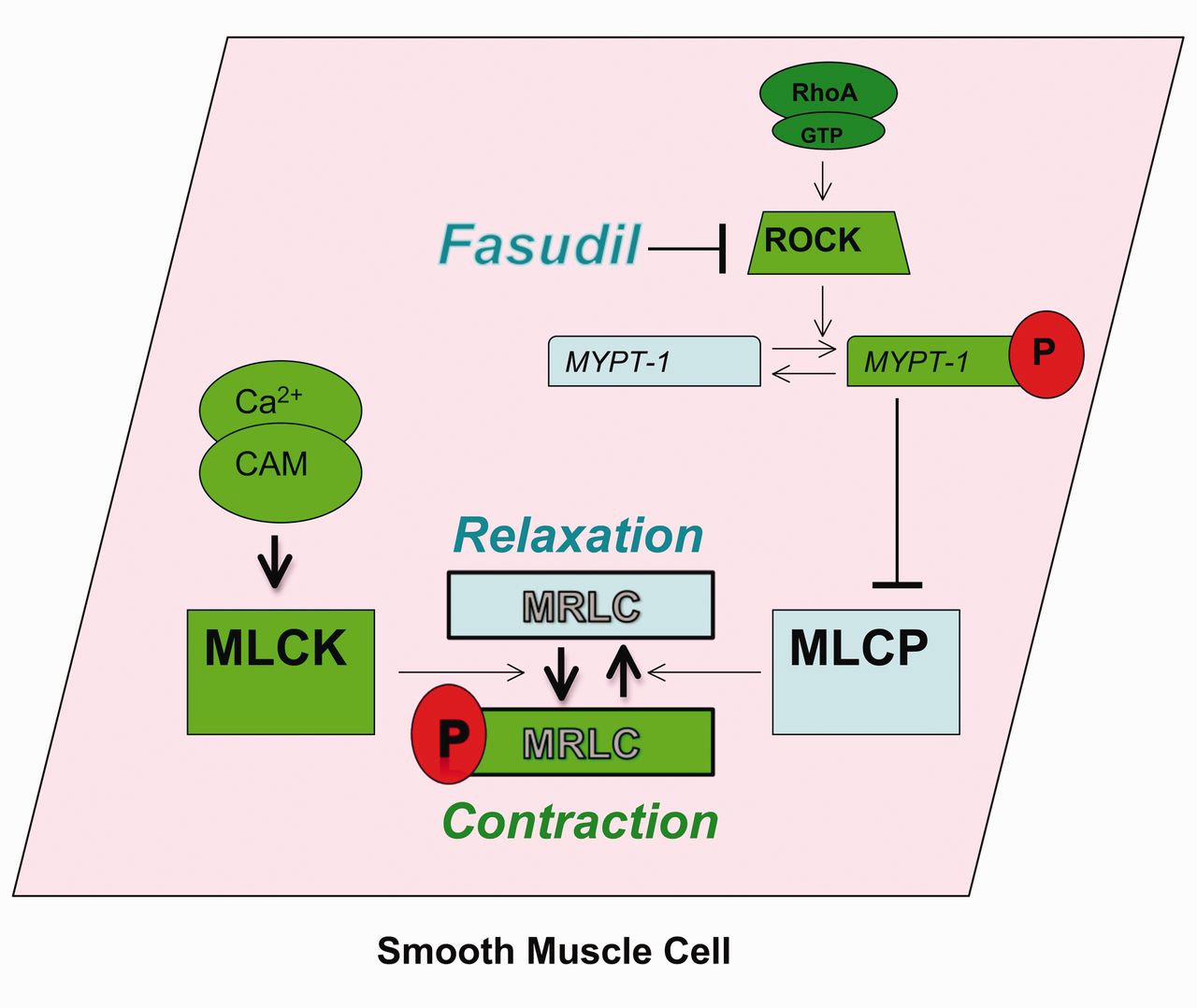
The mechanism of the rho kinase inhibitor, fasudil. RhoA is a small G-protein that activates downstream effectors, including the rho kinases (ROCKs). One target of ROCK is myosin phosphatase target subunit (MYPT-1), which is the regulatory subunit of myosin light chain (MLCP). Phosphorylation (P) of MYPT-1 results in MLCP inhibition, decreased MLC dephosphorylation, and increased smooth-muscle cell contraction, at any given Ca2+ concentration. By inhibiting ROCK, fasudil blocks this signaling. GTP = guanosine-5′-triphosphate. CAM = calmodulin. MLC = myosin light chain. MLCP = myosin light chain phosphatase. MLCK = myosin light chain kinase.
Clinical data, although limited, have been less impressive. Fujita and colleagues administered fasudil, a Rho kinase inhibitor, and INO to 15 adult patients with PAH.226 In that study, the effects of fasudil and INO were similar: there was a decrease in PAP but not PVR. Li and colleagues investigated the acute effects of an intravenous infusion of fasudil in 12 children (mean age 12.3 years) with PAH associated with congenital heart disease.227 Fasudil decreased PAP and PVR, and increased cardiac output and mixed venous oxygen saturation. Comparisons to other vasodilators were not made, however. Likewise, Ishikura and colleagues administered fasudil via intravenous infusion to 8 adult patients with PAH, and found a decrease in PVR and mean PAP, and increased cardiac output.228 Finally, Fukumoto and colleagues administered fasudil to 9 adult patients with PH.229 Some patients were receiving oral or intravenous PGI2 at the time of study. The patients underwent right-heart catheterization, which revealed decreased PVR without a significant change in PAP or cardiac output. Together these studies suggest potential efficacy for Rho kinase inhibition in the treatment of PH, but large controlled trials are needed.
Support of Right-Ventricular Function
Mortality from PH is most directly related to right-ventricular function. The therapies outlined above may improve right-ventricular function to the extent that they decrease right-ventricular afterload, although emerging data suggest that some of these therapies, such as PDE5 inhibition and ET-1 receptor antagonism, may also enhance or impair (respectively) contractility of the hypertrophied right ventricle.113,230 However, in addition to afterload reduction, other therapies that support the right ventricle may be necessary, especially in acute care settings.
Under conditions of increased afterload, the contractility of right-ventricular cardiomyocytes increases initially, due to changes in sarcomere length/tension relationships, increased Ca+2 sensitivity, and alterations in force/frequency relationships.231,232 In addition, the time course over which right-ventricular afterload increases and the state of the right ventricle (in particular, right-ventricular mass) influences the degree to which the right ventricle can compensate.233 For example, patients with Eisenmenger syndrome tolerate elevated right-ventricular afterload far better than patients with normal right ventricles who suffer an acute pulmonary embolism.231,234
Nonetheless, over some period of time (acutely or chronically) compensatory mechanisms fail, leading to elevations in right-ventricular end-diastolic volume and decreased output. Due to ventricular interdependence, increases in right-ventricular end-diastolic volume result directly in decreased left-ventricular filling and decreased systemic output.235 In fact, diastolic ventricular interactions, with decreases in left-ventricular end-diastolic volume, are more closely related to stroke volume than PAP in patients with PAH.236 It is also important to recognize that right and left-ventricular contractility are directly related. The ventricles share muscle fibers, the interventricular septum, and the pericardial space. Based on studies that used electrically-isolated right-heart preparations and experimental aortic constriction, it is estimated that 20–40% of right-ventricular systolic pressure is due to left-ventricular contraction.237–239 In addition, right coronary artery perfusion is dependent, in large part, on the pressure gradient between the aortic root and right ventricle.
Taken together, then, the principles of right-ventricular support are: reduce right-ventricular afterload (ie, reduce pulmonary vascular impedance); optimize right-ventricular volume; augment right-ventricular contractility; and maintain left-ventricular contractility and systemic vascular resistance. Importantly, this strategy requires adequate left-ventricular function. The physiology associated with PH due to left-heart failure is quite different. Left-heart failure is associated with elevated left-ventricular end-diastolic volume and pressure, which is the reverse situation of right-heart failure due to PAH. Moreover, in this situation, decreased right-ventricular afterload and/or increased systemic vascular resistance could result in clinical deterioration, with pulmonary edema or impaired cardiac output.16,17,240 Interestingly, however, sildenafil has been shown to increase cardiac output in patients with PH secondary to left-heart failure, presumably due to reductions in pulmonary and systemic vascular resistance.241,242
The optimization of right-ventricular volume presents a substantial clinical challenge, as the proper management is dependent on the particular situation.243–248 Although volume loading may be necessary in some situations, excessive volume may provoke adverse diastolic ventricular interactions. Management aimed at decreasing right-ventricular volume (eg, diuretics) may be necessary.248,249
Inotropes are often necessary in order to augment right-ventricular contractility, but it remains unclear if one agent is superior. Although dopamine has been shown to increase cardiac output in patients with PH, Liet and colleagues found that dopamine increased the ratio of PVR to systemic vascular resistance in preterm infants who had a widely patent ductus arteriosus.250,251 Based on animal studies, epinephrine may have a superior hemodynamic profile in the setting of PH, compared to dopamine, including a decrease in the PVR to systemic vascular resistance ratio, but direct clinical evidence is sparse.252 Low-dose dobutamine may reduce PVR and increase right-ventricular contractility. Several clinical studies have demonstrated the efficacy of dobutamine in adult patients with PH.253–255 Likewise, milrinone, a PDE3 inhibitor and inodilator that augments ventricular contractility while decreasing PVR and systemic vascular resistance, improves right-ventricular output in adult patients with PH.256–258 The decrease in systemic vascular resistance may not be desirable and thus may need to be addressed by the addition of a vasopressor. Finally, the drug levosimendan, which is a Ca2+ sensitizing agent and PDE3 inhibitor, holds great promise. Levosimendan decreases PVR and improves right-ventricular output in adult patients with right-ventricular failure secondary to several conditions, including PH.259–263
The role of vasopressors is to increase systemic vascular resistance to augment right-ventricular output by increasing left-ventricular systolic pressure, and to maintain right coronary perfusion. Norepinephrine has been validated as a useful agent in several animal studies.264,265 Tourneux and colleagues demonstrated that norepinephrine increased left-ventricular output, systemic arterial pressure, and pulmonary blood flow, while decreasing the ratio of pulmonary to systemic pressure in 18 newborns with PPHN.266 Phenylephrine increases right coronary blood flow in the setting of increased right-ventricular pressure, but may also increase PVR.267,268 Vasopressin, a systemic vasoconstrictor and pulmonary vasodilator, has been advocated in the treatment of right-ventricular failure secondary to PH, with several positive clinical studies.269–274
Finally, atrial septostomy as a part of the management of chronic PH has been advocated to allow for decompression of the right ventricle due to right-to-left shunting.262,275–280 Severe hypoxemia with this approach remains a concern. Recently, Labombarda and colleagues described favorable results with the placement of a Potts anastomosis (descending aorta to left pulmonary artery) in 2 children with severe idiopathic PH, thereby directing desaturated blood to the lower body.281
Treatment of Underlying Disease
The ability to impact the course of PH by treating associated conditions is highly variable. Early repair of congenital cardiac defects represents the most successful effort to alter the natural history of PH.34,87,282,283 Likewise, PH related to treatable left-heart disease would be expected to resolve in most cases, depending on the timing of the repair. However, treatment for other associated conditions may not decrease the incidence of PH. For example, PH can develop with schistosomiasis and HIV infection, despite treatment.284,285 The reversal of PH associated with portal hypertension after liver transplant has been described, but not in a large series.286,287 Likewise, the reversal of PH associated with systemic lupus erythematosus after hematopoietic stem-cell transplantation has been described, but only in case reports.288 Steroids have been successful in treating some patients with autoimmune disease, mixed connective-tissue disease, POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes) syndrome, Langerhans cell granulomatosis, and sarcoidosis.289–293 Advances in the management of sickle-cell disease may decrease the incidence of associated PH, but definitive studies are lacking.294
Subsets of newborns with PPHN are often treatable, and can ultimately survive without PH.295 Several reports have described the reversal of PH after tonsillectomy or adenoidectomy for the treatment of obstructive sleep apnea.296–298 In addition, PH related to high altitude can be reversed when the patient moves to sea level.299 Home oxygen therapy is a relatively common treatment for pediatric patients with PH or at risk for developing PH, but the data conflict about whether oxygen therapy alters the disease course, probably due to differences between the diseases that have been studied.300,301
Finally, an increasing number of metabolic conditions have been found to be associated with PH. For example, the association between thyroid disorders and PH is now well established and, in fact, therapy has been shown to reverse PH in these patients.302
Promotion of Regressive Remodeling of Structural Pulmonary Vascular Changes
Right-heart failure due to elevated pulmonary vascular impedance is the ultimate cause of mortality in most patients with PH. The majority of patients with advanced disease do not respond to acute pulmonary vasodilators, and yet most available therapies either augment pathways that cause vasodilation or inhibit pathways that cause vasoconstriction. Taken together, it can be seen that an approach aimed at promoting the regression of structural pulmonary vascular remodeling may be a fundamentally more effective paradigm for patients with PH not associated with treatable conditions or with advanced PH. Fortunately, although such therapies are unavailable currently, they may be on the horizon.
Recent insights into the mechanisms responsible for advanced pulmonary vascular remodeling have demonstrated interesting similarities to neoplasia. Thus, signaling pathways of interest in cancer research have been studied in models of PH. Indeed, pulmonary vascular remodeling is known to involve abnormalities in proliferation, cell migration, and apoptosis. In addition, pulmonary vascular remodeling also involves processes related to inflammation.
Tyrosine Kinase Inhibitors.
An important recent study examined the efficacy of imatinib mesylate, a tyrosine kinase inhibitor, in patients with PH refractory to other therapies.303 Imatinib, which was developed for the treatment of leukemia, inhibits PDGF receptor, which has been implicated in the pathobiology of advanced PAH.63 Imatinib reversed advanced PH in a monocrotaline rat model and a chronic hypoxia mouse model,63 so Ghofrani and colleagues evaluated oral imatinib in a 24-week randomized, double-blind, placebo-controlled phase II study that included 59 adult patients with PAH.303 Imatinib decreased PVR and increased cardiac output compared to placebo. These results are intriguing, although much more work is needed to assess the potential impact on pulmonary vascular remodeling.
Likewise, sorafenib inhibits the serine/threonine kinases Raf-1 and b-Raf, in addition to PDGF receptor, VEGF receptor, c-Kit, and Flt-3.304 Raf-1 is a key downstream effector for mitogen-activated protein kinase signaling, and in this way may participate in the regulation of proliferation and apoptosis. PDGF, VEGF receptors, c-Kit, and Flt-3 are involved in angiogenesis. Thus, sorafenib has potential for the treatment of vascular remodeling in PH. Indeed, in an experimental rat monocrotaline model, sorafenib improved hemodynamics, reversed pulmonary vascular remodeling, and decreased right-ventricular mass to a greater extent than both imatinib and vehicle control.305
Survivin.
Survivin is a member of the “inhibitor of apoptosis” family that is ubiquitous in cancer, but not normal, cells.306 Interestingly, McMurtry and colleagues, found that survivin was expressed in the pulmonary arteries of 6 patients with PAH.307 The same study found that survivin was similarly increased in the pulmonary arteries of rats with a monocrotaline-induced model of PH, and that gene therapy with a dominant-negative survivin mutant reversed vascular remodeling, improved hemodynamics, decreased right-ventricular hypertrophy, and prolonged survival.
Statins.
Statins, or HMG-CoA reductase inhibitors, have been found to positively impact a number of pathways in experimental PH. For example, in both monocrotaline and chronic hypoxia rodent models, statins improved pulmonary hemodynamics and vascular remodeling by improving endothelial function, decreasing inflammation, inducing apoptosis, and decreasing neointimal proliferation.308–315 Kao and colleagues performed an open-label observational study that included 16 adult patients with PAH treated with simvastatin.316 Simvastatin appeared to improve exercise capacity and cardiac output while decreasing right-ventricular systolic pressure. Lee and colleagues performed a double-blind trial in 53 adult patients with COPD and PH, in which patients received pravastatin or placebo for 6 months.317 Pravastatin treatment was associated with improvements in exercise capacity and Borg dyspnea score. Systolic PAP decreased in the treatment group. Interestingly, urinary excretion of ET-1 decreased in the treatment group and was correlated with improvements in exercise capacity. These improvements in exercise capacity were not replicated in a more recent trial by Wilkins and colleagues that included 42 patients with PAH, treated with simvastatin for 6–12 months.318 However, the primary outcome in that study was right-ventricular mass measured via cardiac magnetic resonance imaging. Simvastatin initially reduced right-ventricular mass at 6 months in the treatment group, but the effect was not sustained at 12 months. King and Day recently conducted an observational study that included a review of 12 children with PH treated with simvastatin,319 and found no clear benefit from simvastatin.
Peroxisome Proliferator-Activated Receptor Gamma.
Peroxisome proliferator-activated receptors (PPARs), which are members of a nuclear hormone receptor superfamily, are rapidly emerging as integral mediators of a wide array of disease processes, including vascular disorders.320 Although investigations of the vasculature have focused primarily on the systemic circulation, one study showed that PPAR gamma (PPARγ), one of the 3 PPAR subclasses, was decreased in lung tissue taken from patients with severe PAH.321,322 Subsequently, increasing efforts have been made to evaluate PPARγ as a therapy for PH. Indeed, studies have identified interactions between PPARγ and multiple mediators that are important in the pathogenesis of pulmonary vascular disease, including nitric oxide, ET-1, PGI2, ROCK, EPCs, asymmetric dimethylarginine, insulin, oxidative stress, and the BMPR2 pathway.322–331 PPARγ expression was decreased in several studies that used rodent models of PH, and PPARγ agonist treatment attenuated pulmonary vascular remodeling.332–335 In an interesting recent study, Falcetti and colleagues demonstrated that PPARγ expression was enhanced in the medial layer of arteries from patients with idiopathic PH, compared to controls.322 Whether that finding conflicts with the previous description of decreased lung tissue expression in advanced PH is unclear, since the initial study did not quantitate expression in the medial muscle layers.321 Moreover, those investigators demonstrated that the PPARγ agonist rosiglitazone potentiated, and PPARγ antagonism inhibited, the anti-proliferative effects of treprostinil in pulmonary artery smooth-muscle cells, whereas down-regulation of the IP receptor had no effect.
Emerging Therapies.
A number of approaches are emerging that may impact vascular remodeling. Caspases are key regulators of apoptosis and are inhibited by intracellular K+. Voltage-gated K+ channels are decreased in patients with PH.336 Recent, studies found that the transcription factor nuclear factor of activated T cells (NFAT) was increased in experimental PH, and that inhibition of NFAT increased K+ channels, inhibited apoptosis, and reversed the PH.337 Recent studies indicate that the signal transducers and activators of transcription (STAT) protein family have binding sites within the promoter regions of the major NFAT isoforms.337 The isoform STAT3 promotes the expression of the proto-oncogene, Pim1, which has been linked to proliferation and resistance to apoptosis in some cancers. Moreover, a recent study found that Pim1 expression was increased in human PAH and that inhibition of STAT3-Pim1-NFAT signaling by nebulized Pim1 siRNA reversed monocrotaline-induced PAH in rats.338
Changes in the extracellular matrix of pulmonary vascular walls participate in the vascular remodeling in PH. Cowan and colleagues showed that elastase inhibition completely reversed the hemodynamic and structural changes in experimental PH.339 Elastase inhibitors are not available clinically, but inhibitors of downstream mediators, including epidermal growth factor (EGF) receptors, are used clinically to treat cancers. That same group demonstrated that the EGF receptor inhibitor PKI166 attenuated PH in a rat monocrotaline PH model.340
Dichloroacetate is a drug that is used clinically to treat mitochondrial disorders in children.341 Interestingly, dichloroacetate reversed the hemodynamic and structural abnormalities in rodent PH models, in part through an activation of mitochondrial-dependent apoptosis.342,343
Lung Transplantation
Isolated lung transplantation, as opposed to requisite heart and lung transplantation, is now a viable option for children with end-staged PH. According to the registry of the International Society of Heart and Lung Transplantation, 173 patients less than 8 years of age underwent lung transplantation for PH between 1990 and 2006.344,345 Survival in children at 3 years and 5 years was reported to be 60% and 50%, respectively, which was similar to that in adults.345 Moreover, this recent report suggested that outcomes were improving. Idiopathic PH, PH associated with congenital heart disease (including Eisenmenger syndrome), and pulmonary vein stenosis or pulmonary veno-occlusive disease are the most common diagnoses in patients who undergo lung transplantation for PH.345 The optimal timing for lung transplantation in children with PH is not clear. A study that included 50 children with PH found that the value of mean right-atrial pressure multiplied by the indexed PVR independently estimated the probability of death at 1 year and 3 years after cardiac catheterization. Those authors suggested that this composite value might be useful for identifying children who should undergo lung transplantation.346
Summary
Over the past several decades, fundamental advances in pulmonary vascular biology have been translated into effective therapies that have improved the quality of life and prolonged survival for neonates, infants, and children with PH. The challenge remains to better characterize the differences between the clinical conditions that cause PH, in order to devise and tailor specific, more effective therapies. Among clinicians and researchers in the field of PH it is increasingly recognized that therapies other than endothelial-based vasodilators are needed. Furthermore, it is recognized that large gaps exist between the available preclinical animal models and clinical disease. For example, many experimental therapies have been shown to reverse monocrotaline-induced PAH in rodents, but none of these therapies have had similar effects on advanced structural disease in humans.347 In addition, the hemodynamic complexities associated with PH are now better understood, which will result in the more routine consideration of indices such as vascular shear stress and pulmonary vascular compliance, in addition to measurements of PAP and calculated PVR. Likewise, alterations in these hemodynamic forces are not uniform throughout the pulmonary vasculature, probably differing in important ways, for example, between large proximal arteries and distal arterioles. Technological advances in imaging and improved computational modeling are emerging to address these sorts of considerations. Further research will elucidate the mechanisms responsible for structural remodeling, hopefully with the development of therapies that promote the regression of advanced structural disease. These studies are likely to make use of multiple preclinical animal models (including models of advanced structural disease), with more sophisticated determinations of cardiopulmonary hemodynamics and morphometric analysis. A better understanding of the genetic underpinnings of PH will hopefully lead to the ability to tailor therapy for particular patients and etiologies. It remains to be seen to what extent these advances will contribute to a unifying theory of PH as opposed to a fragmentation of our current conception of PH into distinct novel disease processes.
Discussion
Willson:
It's like a poster for translational research: clinical care follows the biology, and it's very exciting. After Flolan [epoprostenol], what's next in the kids that you deal with, and how do you advance therapy in these kids if they don't respond at one level?
Fineman:
We discuss that in the clinic all the time. Some of us are going with the approach that if they've got bad disease and are not responding, we go after all 3 cascades up front: prostanoid, NO-cGMP, and ET-1. So we'll add sildenafil or tadalafil to augment cGMP, and bosentan to block ET receptors. The issue with bosentan is that about 5% of the time the liver-function test results become elevated and you have to stop it, so it requires blood draws once a month, which can be an important issue in a pediatric patient.
Unfortunately, there are no data to drive these decisions. Hopefully, future research will identify the relative roles of different pathobiologies in these patients, allowing us to tailor our choices based on the underlying pathobiology. It is becoming clear that although the end-stage anatomic changes may be similar between patients, the underlying pathobiology may be very different. This is clearly a spectrum of disease.
Branson:
Is there ever a reason to have a patient on mechanical ventilation getting INO and aerosolized Flolan? I see it happening all the time, and it's an issue because the aerosolized Flolan fouls the sample line on the machine, making it difficult to use them both at the same time.
Fineman:
I could see using both in a patient who is chronically ill on iloprost or inhaled prostacyclin and who gets critically ill. You may want to use INO in that ventilated patient and not stop their prostanoid therapy during that time. Without knowing if they respond particularly well to prostanoids versus INO, in general a good dose of INO is probably going to offset anything you can achieve with prostanoids in the acute setting.
Walsh:
If we initiated INO without a positive response and then added Flolan and received a positive response, should we then consider stopping the INO?
Fineman:
Yes. If it's a chronic problem and if they have a good response to INO as well, then you could argue to have them on sildenafil or tadalafil long-term, to augment that cascade. But if you saw no response to that cascade and you start getting a response with Flolan, then you could argue to just leave them on a prostanoid.
DiBlasi:
At Seattle Children's Hospital we're conducting a study of inhaled iloprost and our ability to wean INO. However, it has been very difficult to enroll patients, because they are receiving HFOV [high-frequency oscillatory ventilation], and it is unclear if we can effectively deliver nebulized iloprost during HFOV. I have searched the literature and there are virtually no data on aerosol drug delivery with the available aerosol delivery devices during HFOV. Luckily, there are a few posters being presented at the AARC [American Association for Respiratory Care] Open Forum this year that might help answer that question. Do you have any evidence that administering this drug—not albuterol, but some of the vasoactive drugs you mentioned in your presentation—are safe and effective during HFOV?
Fineman:
No, I don't know of any studies that have looked at that. It's problematic.
Cheifetz:
Jeff, that was probably the best review of PH I have seen. You presented numerous therapy options. If you use your crystal ball, with everything that is in the pipeline, how do you think chronic pulmonary hypertension will be most optimally managed in 5 years?
Fineman:
We're learning a great deal about the pathobiology of PH, and are beginning to appreciate the spectrum of possible pathobiologies. In particular patients, all the new therapies—the kinase inhibitors, the anti-inflammatory agents, the PPAR gamma agonists, and the NOS couplers—can be beneficial. This has been demonstrated in isolated case reports. The problem is identifying which patients will benefit from which agents. I am concerned that we won't be able to answer any of these questions. PH is a rare disease, and it's really hard to study these patients. It will require a true collaborative multi-national effort.
Willson:
What about anti-coagulation?
Fineman:
Yes, we use it. Short of there being a known underlying thromboembolic disease, there probably are no great data on anti-coagulants. Whether you use aspirin or coumadin often depends on the child's age, activity level, family preference, et cetera. If they have a central line, you'd probably want to be more aggressive with anti-coagulation.
Cheifetz:
When do you start anticoagulation? On the day of acute presentation, or later?
Fineman:
Usually on presentation. However, if they have not been on anti-coagulation therapy and they now have low output, you may want to start therapy to minimize the risk of thrombus formation in the heart.
Footnotes
- Correspondence: Jeffrey R Fineman MD, Division of Pediatric Critical Care Medicine, University of California, San Francisco CA 94143-1346. E-mail: jeff.fineman{at}ucsf.edu.
Dr Fineman presented a version of this paper at the 47th Respiratory Care Journal Conference, “Neonatal and Pediatric Respiratory Care: What Does the Future Hold?” held November 5–7, 2010, in Scottsdale, Arizona.
This research was partly supported by National Institutes of Health grants HD047349 (to Dr Oishi) and HL61284 (to Dr Fineman), and a LeDucq Foundation Transatlantic Network development grant (to Dr Fineman).
The authors have disclosed no conflicts of interest.
- Copyright © 2011 by Daedalus Enterprises Inc.
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.
- 22.
- 23.↵
- 24.↵
- 25.
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.
- 38.
- 39.
- 40.
- 41.↵
- 42.↵
- 43.↵
- 44.
- 45.
- 46.
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.
- 52.
- 53.
- 54.↵
- 55.
- 56.
- 57.
- 58.↵
- 59.↵
- 60.
- 61.↵
- 62.↵
- 63.↵
- 64.
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.
- 72.
- 73.
- 74.
- 75.↵
- 76.↵
- 77.↵
- 78.
- 79.
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.
- 100.↵
- 101.↵
- 102.
- 103.
- 104.↵
- 105.↵
- 106.↵
- 107.
- 108.↵
- 109.↵
- 110.
- 111.
- 112.↵
- 113.↵
- 114.↵
- 115.
- 116.
- 117.↵
- 118.↵
- 119.↵
- 120.
- 121.↵
- 122.↵
- 123.↵
- 124.↵
- 125.↵
- 126.↵
- 127.↵
- 128.↵
- 129.↵
- 130.
- 131.
- 132.↵
- 133.↵
- 134.↵
- 135.↵
- 136.↵
- 137.↵
- 138.
- 139.↵
- 140.↵
- 141.↵
- 142.↵
- 143.↵
- 144.↵
- 145.↵
- 146.↵
- 147.
- 148.↵
- 149.↵
- 150.↵
- 151.↵
- 152.↵
- 153.↵
- 154.↵
- 155.↵
- 156.↵
- 157.↵
- 158.↵
- 159.↵
- 160.↵
- 161.
- 162.↵
- 163.↵
- 164.↵
- 165.
- 166.
- 167.
- 168.
- 169.↵
- 170.↵
- 171.↵
- 172.↵
- 173.
- 174.↵
- 175.↵
- 176.↵
- 177.
- 178.↵
- 179.↵
- 180.↵
- 181.↵
- 182.↵
- 183.
- 184.
- 185.↵
- 186.↵
- 187.↵
- 188.↵
- 189.↵
- 190.↵
- 191.↵
- 192.↵
- 193.↵
- 194.↵
- 195.↵
- 196.↵
- 197.↵
- 198.↵
- 199.↵
- 200.↵
- 201.↵
- 202.
- 203.↵
- 204.↵
- 205.↵
- 206.↵
- 207.
- 208.↵
- 209.↵
- 210.↵
- 211.↵
- 212.↵
- 213.↵
- 214.↵
- 215.↵
- 216.↵
- 217.↵
- 218.↵
- 219.↵
- 220.
- 221.↵
- 222.↵
- 223.↵
- 224.↵
- 225.↵
- 226.↵
- 227.↵
- 228.↵
- 229.↵
- 230.↵
- 231.↵
- 232.↵
- 233.↵
- 234.↵
- 235.↵
- 236.↵
- 237.↵
- 238.
- 239.↵
- 240.↵
- 241.↵
- 242.↵
- 243.↵
- 244.
- 245.
- 246.
- 247.
- 248.↵
- 249.↵
- 250.↵
- 251.↵
- 252.↵
- 253.↵
- 254.
- 255.↵
- 256.↵
- 257.
- 258.↵
- 259.↵
- 260.
- 261.
- 262.↵
- 263.↵
- 264.↵
- 265.↵
- 266.↵
- 267.↵
- 268.↵
- 269.↵
- 270.
- 271.
- 272.
- 273.
- 274.↵
- 275.↵
- 276.
- 277.
- 278.
- 279.
- 280.↵
- 281.↵
- 282.↵
- 283.↵
- 284.↵
- 285.↵
- 286.↵
- 287.↵
- 288.↵
- 289.↵
- 290.
- 291.
- 292.
- 293.↵
- 294.↵
- 295.↵
- 296.↵
- 297.
- 298.↵
- 299.↵
- 300.↵
- 301.↵
- 302.↵
- 303.↵
- 304.↵
- 305.↵
- 306.↵
- 307.↵
- 308.↵
- 309.
- 310.
- 311.
- 312.
- 313.
- 314.
- 315.↵
- 316.↵
- 317.↵
- 318.↵
- 319.↵
- 320.↵
- 321.↵
- 322.↵
- 323.
- 324.
- 325.
- 326.
- 327.
- 328.
- 329.
- 330.
- 331.↵
- 332.↵
- 333.
- 334.
- 335.↵
- 336.↵
- 337.↵
- 338.↵
- 339.↵
- 340.↵
- 341.↵
- 342.↵
- 343.↵
- 344.↵
- 345.↵
- 346.↵
- 347.↵





{kind=link}
{kind=link}
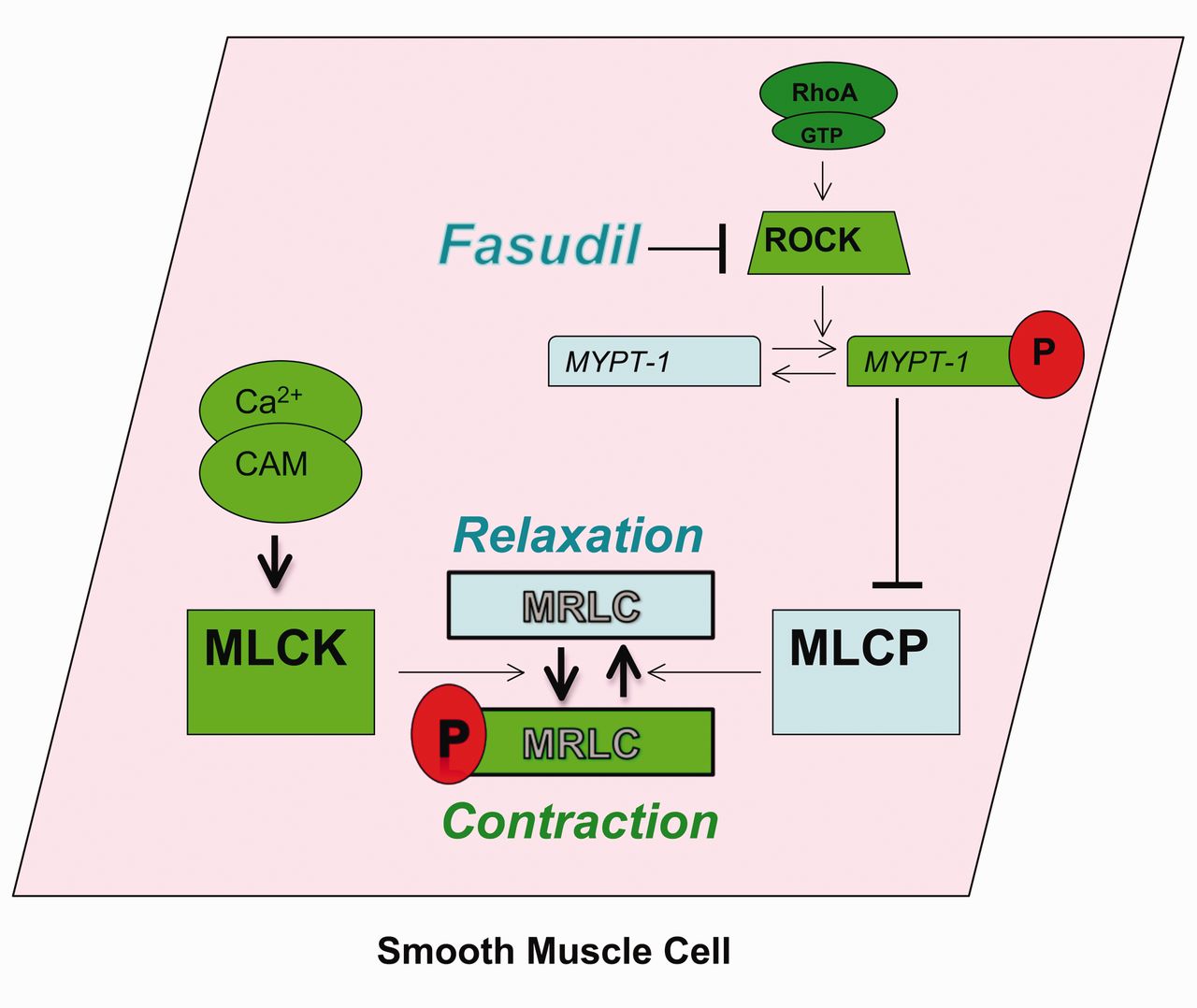{kind=link}