

Abstract
The physiologic changes that occur in ventilation during sleep contribute to nocturnal oxygen desaturation in those with lung disease. Nocturnal supplemental oxygen is often used as therapy, although convincing data exist only for those who are hypoxemic both during sleep and wake. Ongoing trials may help address whether oxygen should be used in those with only desaturation during sleep. If used, oxygen should be dosed as needed, and patients should be monitored for hypercapnia. Because of its prevalence, obstructive sleep apnea may commonly overlap with lung disease in many patients and have important consequences. Patients with overlap syndromes may be good candidates for noninvasive ventilation during sleep.
Introduction
Breathing is tightly controlled by a number of mechanisms to ensure adequate oxygenation, carbon dioxide excretion, and maintenance of acid-base status. However, some mechanisms are altered by the sleep state. Although oxygenation and ventilation remain adequate in those with normal respiratory function, in those with lung disease, sleep can lead to hypoxemia and/or hypercapnia. Furthermore, sleep-disordered breathing (obstructive sleep apnea [OSA] or central sleep apnea) can independently lead to blood gas abnormalities. Here, basic concepts of control of breathing during wake and sleep are reviewed, ultimately with an emphasis on clinical management in those with lung disease (particularly COPD, which has been reasonably well studied) and concomitant sleep-disordered breathing. Nocturnal oxygen is commonly used in those with nocturnal hypoxemia, with little data to guide clinicians.
Control of Breathing During Wakefulness
Breathing arises from the respiratory “pacemaker”: a collection of cells located in the medulla. The output of this pacemaker is subsequently and continuously modified by multiple sources to regulate breathing. These include other central nervous system components, from the pons through the cortex; central and peripheral chemo-receptors; and lung stretch receptors and muscles of respiration, all of which can influence breathing frequency, tidal volume, and inspiratory time. These inputs allow the respiratory system to maintain homeostasis across a wide range of activities, from sleep to maximal exercise, without conscious effort. The overall goal of the respiratory system is to maintain stable levels of oxygen, carbon dioxide, and hydrogen ions in the body. Thus, hypercapnia, acidemia, and hypoxemia are the stimuli that modify ventilation via chemoreceptors. So-called “central” chemo-receptors are located in the medulla and are in direct contact with cerebrospinal fluid, owing to their proximity to the medulla's ventral surface. The central chemoreceptors are separated from blood flow by the semipermeable blood-brain barrier, which allows passage of CO2 but not H+ or HCO3–. Lipid soluble CO2 enters the central nervous system, where it is rapidly hydrated, leading to an increase in the H+ concentration. H+ ions, in turn, stimulate the central chemoreceptors to increase ventilation.1–3 Since there is minimal buffering of CO2 in the cerebrospinal fluid, alveolar ventilation is very sensitive to changes in PaCO2. For example, alveolar minute ventilation increases by 2–3 L/min for each millimeter of mercury rise in PaCO2. After 1–2 days of increased ventilation, the kidneys increase excretion of HCO3– in the urine, which helps to restore the CO2 level in the cerebrospinal fluid and decreases the stimulus for increased ventilation.
Classically, “peripheral” chemoreceptors (located in the carotid bodies at the bifurcation of the common carotid arteries and in the aortic bodies located above the aortic arch) respond to drops in PaO2 by increasing ventilation.4 The effect of hypoxemia is nonlinear, with only mild effects of hypoxemia on nerve activity and ventilation until the PaO2 decreases to < 60 mm Hg, when the effect of hypoxemia becomes more pronounced. These receptors are actually more sensitive to decreases in arterial pH. Traditionally, the central and peripheral chemical controls of ventilation were thought to behave independently, but there is emerging evidence that they are interdependent.5 For example, the sensitivity of the medullary chemoreceptors may be partially determined from the input from the carotid body chemoreceptors. Some data also suggest that the degree of central stimulation can affect the sensitivity of the peripheral chemoreceptors.6 Thus, the prevailing evidence suggests a hyperadditive model, such that the responsiveness of central chemoreceptors can be increased by stimuli at the peripheral chemoreceptors.
In addition to chemoreceptor inputs, breathing is also affected by input from both the limbic system and cortex, by efferent input from muscles of locomotion, and by receptors within the lungs themselves. Cortical inputs allow emotion (eg, anxiety) to affect respiration. During strenuous exercise, O2 consumption and CO2 production can increase 20-fold. These marked changes are matched by an increase in ventilation and cardiac output that keep PaCO2, PaO2, and arterial pH relatively constant. Interestingly, the ventilation increases in parallel with muscle activity and cannot be explained simply by changes in blood gases, since ventilation increases precede any change in PaO2 or PaCO2. The stimulus underlying exercise-induced hyperpnea remains unclear, although emerging evidence supports the hypothesis of feedback from exercising limbs.7,8 In a recent trial, men were injected with lumbar intrathecal fentanyl, which impaired centrally projecting muscle afferents, before being asked to exercise. The injected subjects had significant hypoventilation (4–7 mm Hg rise at peak exercise), compared to subjects injected with saline. The influences of systemic absorption of fentanyl and possible direct medullary effects were excluded. Thus, these results illustrate the importance of muscle afferent feedback to the central nervous system and overall control of breathing.
Finally, in certain disease states, receptors within the lung are important. J or “juxta-capillary” receptors are located in the alveolar walls, adjacent to capillaries. They are stimulated by interstitial fluid volume and can lead to rapid, shallow breathing, associated with dyspnea.9–11 These J receptors are thought to be important in the dyspnea associated with congestive heart failure and may increase ventilatory drive in patients with Cheyne-Stokes breathing. Pulmonary stretch receptors are located in the smooth muscle of large and small airways. The Hering-Breuer inflation reflex is triggered to prevent overinflation of the lungs. It is activated only at large tidal volumes (> 1,000 mL).12 When the pulmonary stretch receptors are stretched, they send impulses via the vagus nerve to the brainstem, which responds by increasing expiratory time and decreasing breathing frequency.13,14 The opposite is also true: the Hering-Breuer deflation reflex describes how sudden lung collapse initiates inspiratory activity.
Changes in Respiration During Sleep
The same basic mechanisms active during wakefulness are also relevant during sleep. Importantly, however, there is a lack of behavioral control, and the responses of specific mechanisms are altered during sleep and during the different sleep stages. The major changes are blunted ventilatory response to hypoxia and hypercapnia (Fig. 1).15–17 These changes lead to a higher CO2 set point: approximately 45 mm Hg, as opposed to 40 mm Hg during wakefulness. Thus, alveolar ventilation is reduced during sleep, compared to wakefulness. Most of the drop in minute ventilation is due to a decrease in tidal volume that is not fully compensated for by a concomitant increase in breathing frequency. Additionally, there are decreased resistance load responses; that is, the ability to respond to increments in the work of breathing is reduced.18 This finding is important, given the normal narrowing of the upper airway that occurs during sleep, even without OSA.19 During wakefulness, pharyngeal muscle activity is maintained by reflex-driven pharyngeal dilator muscle activity. At sleep onset there is reduced reflex muscle activation and wakefulness-modulated excitatory output to the upper airway musculature.20 Thus, a number of factors contribute to reduced ventilation during sleep.

Control of ventilation during sleep. The hypercapnic respiratory response, the major determinant of ventilation, decreases from wakefulness to sleep, and falls further during rapid-eye-movement (REM) sleep. (From Reference 17, with permission.)
All of the above-mentioned alterations that tend to compromise alveolar ventilation are most affected during rapid eye movement (REM) sleep. This sleep stage is characterized by skeletal muscle atonia, except for the diaphragm, and shallow, irregular breathing. Thus, ventilation is even further compromised during REM sleep, and upper-airway resistance is even higher than during non-REM sleep. Although less well studied, there are also important hemodynamic changes that occur with sleep state changes, but that seem to be independent of changes in oxygen saturation or PCO2.21 In addition to other cardiovascular changes during REM sleep, pulmonary artery pressures increase out of proportion to blood gas abnormalities.
Sleep and Lung Disease
Nocturnal Oxygen Desaturation
In normal subjects, even with these alterations in ventilation, there are not usually substantial reductions in oxygen saturation or hypercapnia. However, with lung disease, the changes in ventilation mentioned above lead to nocturnal oxygen desaturation (NOD).22,23 The definition of NOD is somewhat arbitrary, but is usually described in terms of oxygen nadir or time below some oxygen saturation limit, such as 88% or 90%. NOD is most well studied within the context of COPD, and appears to be common. Various studies have reported that 27–70% of patients with COPD with awake oxygen saturation of 90–95% experience substantial desaturation at night, particularly during REM sleep (Fig. 2).24–27 In the most recent study (in which patients were treated according to current guidelines), 38% of COPD subjects with a daytime PaO2 of 56–69 mm Hg (mean 65 mm Hg) spent ≥ 30% of the night with an oxygen saturation < 90%.28 In those with severe COPD, the desaturation nadir during sleep is more profound even than during exercise, with oxygen saturation falling an average of 6 ± 3.6% during peak exercise and 13.1 ± 8.9% during sleep.29 Both awake oxygen saturation and daytime arterial carbon dioxide level predict NOD, although imperfectly.30,31

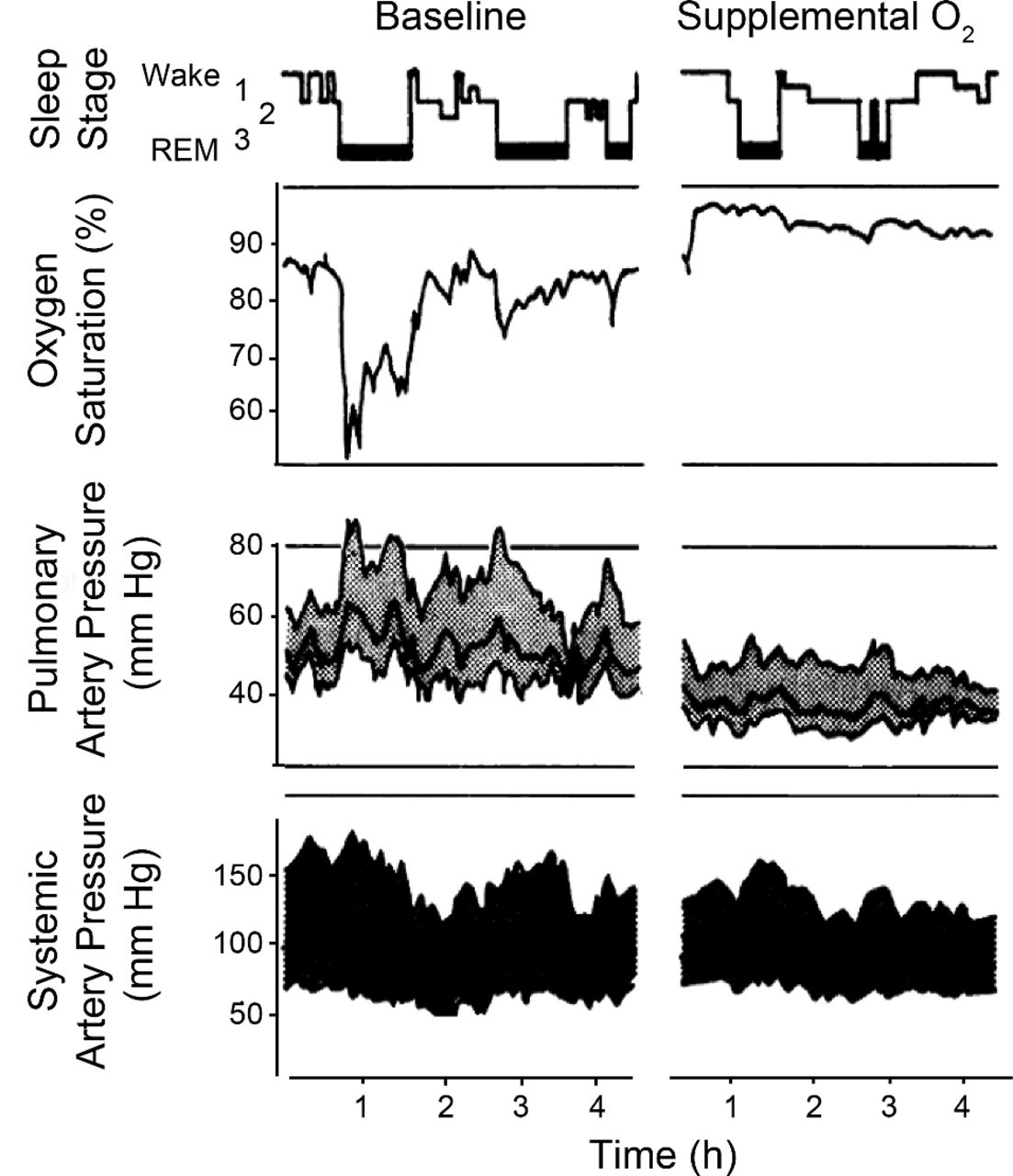
Example of nocturnal oxygen desaturation (NOD) and its physiological consequences. Sleep stage (rapid eye movement [REM] in bold) and transcutaneous oxygen measurements during sleep in a subject with COPD. Hypoxemia worsens during sleep, most substantially during periods of REM sleep. During periods of NOD there are increases in pulmonary and systemic blood pressure. Administration of supplemental oxygen improves NOD and prevents increases in pressure. (From Reference 24, with permission.)
There are probably 3 mechanisms that contribute to NOD. First, as above, there is alveolar hypoventilation, which likely accounts for most of the oxygen desaturation. Becker and colleagues measured minute ventilation during wakefulness, non-REM, and REM sleep in normal patients and those with COPD. In normal subjects, minute ventilation decreased slightly, whereas minute ventilation in COPD patients falls approximately 16% from wakefulness to non-REM sleep, and almost 32% during REM sleep, compared to wakefulness, largely as a result of decreased tidal volume.32 The greater drop in minute ventilation in subjects with COPD may reflect increased dependence on accessory muscles that become hypotonic during sleep, particularly during REM. An alternative explanation comes from the work by O'Donoghue and colleagues, who found an even greater drop in minute ventilation during non-REM sleep in hypercapnic COPD patients.33 When the inspired air was changed to a helium-oxygen mixture (heliox) that should relieve some of the resistance to flow, minute ventilation remained the same, suggesting that the minute ventilation set-point has changed in COPD patients. Hypoventilation cannot explain all of the observed desaturation, since, while there appears to be a uniform rise in carbon dioxide, the partial pressure of oxygen falls a variable amount. This observation suggests that, although never directly measured, ventilation and perfusion matching is altered during sleep, perhaps due to changes in lung volume that occur with sleep onset and/or REM sleep; however, reports are conflicting on the magnitude of any potential lung volume change.33–35
The changes in other lung diseases with sleep have not been as thoroughly studied; however, at least some of these mechanisms likely contribute to NOD in other pulmonary diseases. Patients with clinically important pulmonary disease may have borderline oxygen saturation during the day. Given the shape of the hemoglobin oxygen binding curve, small changes in the partial pressure of oxygen can lead to substantial changes in oxygen saturation. Thus, it is likely that any patient with pulmonary disease with borderline oxygen saturation during the day will have some degree of NOD during the night. For example, Perin and colleagues reported that NOD was common in adult cystic fibrosis patients, especially those with daytime oxygen saturation < 94%, although the oxygen nadir is quite variable even for the same awake oxygen saturation.36
Physiological Consequences of Nocturnal Oxygen Desaturation
Oxygen desaturation has been reported to lead to various cardiac arrhythmias.36 Some studies have also reported that arousals from sleep may be related to episodes of desaturation, and in this way NOD may lead to sleep fragmentation.37 Importantly, NOD causes surges in both systemic and pulmonary blood pressure, which has been of interest when exploring links between OSA and pulmonary and systemic hypertension.24 In OSA patients without intrinsic lung disease, repetitive, transient oxygen desaturation can lead to (generally mild) pulmonary hypertension.38
Interest in the physiological changes detailed above is also driven by 2 clinical observations: that survival is better for patients without NOD, compared to those with NOD, even if daytime oxygen saturation is similar39; and that COPD patients are reported to die more frequently at night than expected. McNicholas and Fitzgerald reported in 1984 that patients admitted to the hospital with chronic bronchitis or emphysema were more likely to die at night than other admitted hospital patients.40 Presumably, the increased rate of nocturnal deaths is due to the effects of hypoxemia on the cardiovascular system (since COPD patients die more frequently from cardiovascular disease than respiratory failure41), but no clear mechanism has been elucidated.
In fact, whether nocturnal hypoxemia alone due to COPD can lead to substantial pulmonary hypertension is controversial. We might expect that, like those with OSA, NOD could lead to pulmonary hypertension over time, especially in those COPD patients with emphysematous changes and capillary destruction that might also lead to pulmonary hypertension. Fletcher and colleagues did find that subjects without substantial daytime hypoxemia but with NOD did have slightly higher pulmonary pressures.42 However, in this instance, NOD may simply have been a marker of more severe disease, since the baseline characteristics between those with NOD and those without were not entirely similar. Subsequent prospective studies by Chaouat and colleagues have not found NOD alone to lead to pulmonary hypertension, or by itself to increase mortality.25,43
In general, other pulmonary diseases such as fibrosis have not been as well studied during sleep.44 The prevalence, mechanisms, and consequences of NOD may be different than with COPD. One pulmonary disease that deserves mention, however, is sickle cell disease. Hypoxemia may contribute to sickle formation and acute chest and painful crises. NOD has been associated with the number of painful crises in children, and was predictive of central nervous system events such as stroke and transient ischemic attack.45,46
Sleep Deprivation and Lung Disease
Many pulmonary diseases impact and have interactions with sleep. The classic example is nocturnal asthma. Salter, in his treatise “On Asthma,” even recommended strong coffee to put off sleep and prevent an asthma attack, since he believed that “Sleep favors asthma.”47,48 The nocturnal decline in peak expiratory flow in asthma is also well documented, although the mechanisms are still unclear, and appears to be very common.49 Sleep quality is often poor in those with nocturnal asthma, and there are daytime consequences on attention and concentration (which improve when nocturnal symptoms are treated).50,51 Similarly, subjective sleep quality is poor in other respiratory diseases, such as COPD and pulmonary fibrosis, with some studies also confirming objectively worse sleep, based on total sleep time or sleep fragmentation.52–55 Thus, lung disease can predispose to acute and chronic sleep deprivation.
Interestingly, the converse may also be true: sleep deprivation may deleteriously affect respiratory function. Some reports suggest that acute sleep deprivation can reduce the hypercapnic ventilatory response.56 In those with COPD, acute sleep deprivation may lead to small reductions in spirometry.57 One can then imagine a vicious cycle in which worsening respiratory disease leads to sleep deprivation, which then causes a further worsening of respiratory function. Sleep deprivation also increases the arousal threshold: a greater stimulus is required to wake up. In those with lung diseases such as asthma or COPD, one stimulus to wake is airway resistance. Here, again, sleep deprivation may have adverse clinical consequences: patients may be less aware of worsening airway resistance during an asthma attack or COPD exacerbation, and wake up too late to call for help or seek treatment. Sleep deprivation and decreased awareness of declining respiratory function may help explain the association between pulmonary disease and deaths during the night.40,58,59 Further study is required in this area.
Interactions Between Lung Disease and Sleep Apnea: The Overlap Syndromes
David Flenley coined the term “overlap syndrome” for the co-existence of OSA and COPD in the same individual.60 However, he allowed for the possibility that any respiratory disease (eg, idiopathic pulmonary fibrosis) could “overlap” with OSA in the same individual (Fig. 3). The most well studied of the overlap syndromes, and the one that Flenley focused on, is that of COPD and OSA. However, there has been an increasing focus looking at OSA in a variety of pulmonary diseases, including idiopathic pulmonary fibrosis, pulmonary hypertension, asthma, and sickle cell disease. In part this reflects the increasing prevalence of OSA in the general population, but may also reflect the use of corticosteroids to manage many pulmonary diseases. Corticosteroids might contribute to OSA through weight gain, or perhaps more directly by theoretically causing an upper airway muscle myopathy, or some other mechanism.

Obstructive Sleep Apnea (OSA) may overlap with any number of pulmonary diseases, including COPD, cystic fibrosis (CF), etc. This overlap will lead to greater and more prolonged NOD, and may contribute to increased morbidity and mortality than either of the constitutive diseases. IPF = interstitial pulmonary fibrosis. (From Reference 60, with permission.)
COPD and OSA: “The” Overlap Syndrome
The overlap syndrome is probably a common and under-recognized clinical entity, in part due to lack of physician awareness and because it has never been more rigorously defined. (The definitions and criteria for both COPD and OSA have also changed over time.) For example, because both COPD and OSA occur on a spectrum of severity, it is unclear at what level of severity the combined diseases begin to have additive or synergistic clinical relevance. It is also unknown if patients with severe COPD and mild OSA should be evaluated and treated similarly to those with mild COPD and severe OSA. Regardless, given the high prevalence of both COPD and OSA, we would expect a large cohort of patients affected with both of these common diseases. Most early studies examined patients with one of these disorders to see how many were also affected with the other illness. These initial studies were not true cross-sectional studies and tended to suggest that the prevalence of the overlap syndrome was very high, suggesting a link between COPD and OSA.61–63 More recent data from the Sleep Heart Health Study and the European MONICA II study did not find an association between the 2 diseases.64,65 The major limitation of these studies is that most subjects had very mild airway obstruction on spirometry. Whether any pathophysiological link exists between OSA and severe COPD is still unknown. It is also still worth emphasizing that, although there may be no increased association between relatively mild COPD and OSA, because of the rising prevalence of these diseases, a patient with either disorder alone will still often have the other disease by chance alone. For example, in the Sleep Heart Health Study and the MONICA II study, GOLD stage II COPD was found in 19% and 11% of subjects, respectively. Sleep-disordered breathing was seen in 14% of subjects in Sleep Heart Health Study (respiratory disturbance index > 15 events/h) and 11% of subjects (apnea-hypopnea index > 5 events/h and excessive daytime sleepiness) in the MONICA II cohort. Thus, even by chance alone, a patient with one disorder has a greater than 10% chance of also having the other disease. When evaluating a patient with one disorder, it is reasonable to screen for the other, at least based on history and review of systems.
Patients with both COPD and OSA have 2 reasons to have NOD, and indeed patients with both OSA and obstructive lung disease have more prolonged oxygen desaturation during the night than those with OSA without obstructive lung disease; the degree of obstruction, as measured by FEV1/FVC, correlates with the risk of prolonged hypoxemia.64 More prolonged hypoxemia, or the coexistence of COPD, appears to increase morbidity and mortality considerably, compared to OSA alone. Patients with the overlap syndrome appear to be at greater risk of pulmonary hypertension and cor pulmonale than patients with either disease alone.66–73 For example, Hawrylkiewicz and colleagues observed that 16% of those with OSA had pulmonary hypertension, compared with 86% of those with overlap syndrome.74 Several pulmonary parameters have been shown to increase mortality in patients with OSA. Both the diagnosis of concomitant COPD and markers of COPD, such as a reduced FEV1 or smoking history, have been shown to be markers for increased mortality in OSA patients.75–77 The largest analysis, by Lavie et al, showed that, in a univariate analysis, COPD conferred a 7-fold risk of death in OSA patients. Conversely, comorbid OSA has recently been reported to increase mortality in patients with COPD. Marin and colleagues have recently published outcomes data on patients with COPD, and those with the overlap syndrome, both with and without CPAP treatment.78 Subjects were initially referred to a sleep clinic for suspicion of sleep-disordered breathing (usually snoring), and then underwent a diagnostic polysomnogram (PSG) and spirometry. After a median follow-up of over 9 years, all-cause mortality was markedly higher in the untreated (no CPAP) overlap group (42.2%), compared to the COPD only group (24.2%). Even when adjusted for COPD severity, comorbid OSA remained a risk factor for death (Fig. 4).

Kaplan-Meier survival curves for outcomes among COPD patients, overlap patients on CPAP, and overlap patients not on CPAP. CPAP treatment was not randomly assigned, a source of potential bias, although markers of disease severity were similar, on average, in the treated and untreated groups. OSA = obstructive sleep apnea. (From Reference 78, with permission.)
The exact mechanism(s) that account for this increased morbidity and mortality risk are not known. Increased risk of death may be due to more prolonged hypoxemia. However, the degree of hypercapnia is likely also greater during the night in overlap syndrome than in either disorder alone, and could also be important. Of concern, like patients with COPD alone, OSA patients have also been shown to die (usually of cardiovascular disease) disproportionately during the night, compared to control groups, who are at greatest risk during the morning hours.79 More recent research has focused on the systemic consequences of COPD and OSA. In addition to the oxidative stress they create, both are associated with inflammation and elevations of a variety of inflammatory mediators (tumor necrosis factor alpha, interleukin 6 and 8). Whether these mechanisms are additive or synergistic is not known.80 In the observational study by Marin, death in the untreated overlap group was most commonly attributed to cardiovascular disease.78 One other intriguing possibility, also reported in this study, is that OSA may contribute to an increased incidence of COPD exacerbations. Exacerbations may accelerate lung function decline and are associated with greater mortality.81,82
The diagnosis of overlap syndrome may also be clinically relevant, since patients with overlap syndrome also have significantly worse quality of life (using the St George's Respiratory Questionnaire), when compared to COPD only controls.83 Of note, the overlap syndrome patients in this study were COPD patients with habitual snoring, but without reported excessive daytime sleepiness or elevated Epworth Sleepiness Scale, who underwent PSG. Of these snoring but non-sleepy COPD patients, two thirds were determined to have an apnea-hypopnea index > 5 events per hour, highlighting the limitations of screening based on symptoms alone. Whether quality of life improves, and by how much, with treatment of OSA is not known, but could be one important consideration in diagnosis and treatment.
Other Overlap Syndromes
Other overlap syndromes, involving OSA and asthma, pulmonary fibrosis, or sickle cell disease, have been less well studied to date. However, OSA is often comorbid with these diseases, and may impact disease severity. The overlap between OSA and asthma is an illustrative example. There is increasing evidence that OSA and asthma commonly occur together, more frequently than would be expected by chance alone. Subjectively, asthma patients frequently report symptoms consistent with OSA, such as snoring and excessive daytime sleepiness.84 In fact, some reported nocturnal asthma attacks may actually represent symptoms of OSA.85 When examined using PSG, those with difficult to treat asthma have had very high rates (> 85%) of concomitant OSA, and OSA appears to be a risk factor for asthma exacerbations.86,87 Mechanistic links between OSA and asthma have been minimally studied to date. Treatment of concomitant OSA is reported to have variable effects on asthma severity. A small study showed that in patients with severe asthma and OSA, application of CPAP did reduce nocturnal worsening of asthma.88 In less severe asthmatics no change was seen in FEV1 or the provocational concentration of a bronchial agonist causing a 20% decrease in FEV1; however, asthma quality of life scores improved a clinically important amount, suggesting unmeasured improvements in lung function during sleep.89,90 Ongoing studies may help answer questions about mechanism and the role that treatment has on modifying asthma.
Although data are sparse, OSA appears common in other types of lung disease and it may contribute to morbidity and mortality. For example, OSA is reported to be very prevalent in patients also diagnosed with idiopathic pulmonary fibrosis, and followed in a specialty fibrosis clinic.91 In selected populations of children with sickle cell disease, OSA is also common, and these children have more prolonged and profound periods of hypoxemia and hypercapnia, compared to OSA-only controls.92 Unlike COPD, the role of CPAP has yet to be defined in these other overlap syndromes. Some authors have hypothesized that CPAP therapy might be difficult to apply in patients with idiopathic pulmonary fibrosis, for example, due to the prevalence of nocturnal dry cough as part of that disease.93
Diagnosis
Prior to diagnostic testing, history and physical examination may be suggestive of concomitant OSA in those with pulmonary disease, and may influence the choice of test. For example, in an adult out-patient pulmonary clinic for patients with asthma or COPD, a well validated questionnaire for OSA incorporating patient symptoms and physical examination findings (the Berlin Questionnaire) suggested that 55% of patients were at high risk for OSA, and subsequent testing in this high risk group found that almost all did indeed have OSA.94 In patients with severe COPD, poor sleep quality is often reported.37,52–54 However, in those with more mild respiratory disease studied in the Sleep Heart Health Study, there were only very minor effects of COPD on sleep quality and architecture.64 Thus, sleep complaints or classic symptoms of OSA in these patients should be evaluated with PSG, although this approach may underestimate the number of affected patients, since some OSA may be minimally symptomatic but still clinically relevant.95 OSA might also be suggested by the presence of pulmonary hypertension that seems out of proportion to the severity of COPD.69,96 Generally, patients with COPD only and pulmonary hypertension will tend to have severe disease and blood-gas abnormalities (FEV1 < 1 L, FEV1/FVC < 50%, and awake partial pressure of oxygen < 55 mm Hg).97 In contrast, patients with COPD and OSA and pulmonary hypertension have less severe intrinsic lung disease (average FEV1 of 1.8 L, FEV1/FVC 64%, and awake arterial partial pressure of oxygen of 64 mm Hg). Based on these data, American Thoracic Society/European Respiratory Society guidelines suggest that those with relatively mild COPD and evidence of pulmonary hypertension should be referred for overnight testing.98 Finally, COPD patients with NOD who develop morning headaches when treated with nocturnal supplemental oxygen likely have OSA and should be referred for PSG.60,99 Of note, when requesting PSG, it may be helpful to alert the registered sleep technician to the diagnosis of lung disease; this may facilitate CPAP titration based on oronasal air flow, rather than on oxygen desaturation.100
For patients not meeting the above criteria, nocturnal oximetry could be considered. While cheaper and more straightforward than PSG, oximetry alone may not distinguish between NOD due to lung disease and NOD due to some combination of lung disease and OSA. Interestingly, although nocturnal oximetry is routinely ordered, physician decisions and management based on nocturnal oximetry results vary greatly.101 In part, this finding reflects the variable definitions of NOD and the paucity of evidence to guide physicians (discussed below). Some have suggested that nocturnal oximetry in those with OSA will have a characteristic cyclical pattern; however, this approach has not yet been well validated.28 In particular, those with predominantly REM-related OSA may be difficult to discern from those with REM-related hypoxemia, and treatment will differ. An alternative approach to overnight oximetry and an in-laboratory attended PSG would be a home sleep test using a portable monitor.102 However, such devices, which measure air flow, respiratory effort, cardiac rate, and oxygen saturation, have never been validated in those with respiratory disorders.103
Testing should probably be undertaken during periods of disease stability.98 Many patients (roughly 33–50%) prescribed oxygen after an acute worsening of disease (eg, COPD exacerbation) no longer meet requirements if re-tested 1–3 months later.104–106 Additionally, as detailed below, optimal medical therapy and/or pulmonary rehabilitation may improve resting oxygen saturation.
Treatment
Oxygen
The role of oxygen therapy has most clearly been defined in those COPD patients with hypoxemia during both wake and sleep by 2 landmark studies: the Nocturnal Oxygen Therapy Trial (NOTT) in the United States, and the Medical Research Council (MRC) trial in the United Kingdom.107,108 In NOTT, subjects with severe daytime hypoxemia (PaO2 < 55 mm Hg or those with PaO2 < 59 mm Hg and evidence of cor pulmonale) were randomized to nocturnal oxygen or continuous ambulatory oxygen. In the MRC trial, subjects with a similar level of hypoxemia (PaO2 40–60 mm Hg) were randomized to sham nocturnal oxygen (air) or nocturnal oxygen. In both cases, mortality was the primary end point. Because of the design of the 2 trials, the results can be aggregated and are often summarized as: some oxygen is better than none at all, and more is better. One interesting difference between the 2 studies was that NOTT recommended that supplemental oxygen be increased by 1 L/min above baseline during sleep or exercise, presumably to counteract NOD. This latter recommendation is implemented with great regional variability around the world,109 and has been the subject of various studies examining the need to increase supplemental oxygen during the night.110,111 At least one study has shown a slightly higher degree of hypercapnia with increasing oxygen supplementation per the NOTT guidelines, although the clinical importance is not known.112,113
Based on the physiological consequences of NOD, and the results of NOTT and MRC trials, it seems intuitive that the treatment for NOD alone (without daytime hypoxemia) should also be supplemental oxygen, and that there should be some measureable impact on health. As shown by Fletcher and Levin, the acute administration of supplemental oxygen can prevent NOD as well as the associated surges in pulmonary and systemic blood pressure (see Fig. 2).24 However, the benefit of long-term nocturnal supplemental oxygen for those with only nocturnal hypoxemia has not been proven. There have been 2 studies in COPD to date. The first, by Fletcher and colleagues, studied COPD patients with only NOD over 3 years, randomized (n = 19 in both groups) to 3 L/min supplemental oxygen at night.114 At the end of the study there was a small decrease in mean pulmonary arterial pressure, compared to baseline, in the treated group (and a slight rise in the untreated group), but there was no difference in mortality. A second, similar study showed no difference in mortality and no difference in pulmonary hemodynamics.115 These results are consistent with those of Gorecka and colleagues, who studied long-term oxygen therapy in patients with more mild hypoxemia (PaO2 56–65 mm Hg).116 Although not measured, their study participants likely had NOD, and there was no survival advantage to supplemental oxygen after 3 years. Overall, correction of nocturnal hypoxemia alone (in patients with daytime normoxia) does not seem to significantly improve pulmonary hemodynamics or mortality, although it may improve sleep quality (based on a single study) and is frequently prescribed.117 For many experts, the use of supplemental oxygen to prevent NOD is still an open question.118,119 At least 2 large multicenter trials are ongoing. The first will specifically look at those with NOD: the International Nocturnal Oxygen Trial (INOX, NCT01044628). Another study of supplemental oxygen in less severe patients will also help answer this question (the Long-Term Oxygen Treatment Trial (LOTT, NCT00692198).120 Until such data are available, for those with NOD in the absence of OSA, supplemental oxygen could be prescribed and patients assessed for symptomatic improvement (in sleep quality, daytime energy, attention, and concentration); therapy should be continued only if there appears to be benefit. As discussed below, supplemental oxygen should be titrated to the minimum level needed to prevent nocturnal hypoxemia.
Can Supplemental Oxygen Cause Harm?
The cumulative data on trials of supplemental oxygen can be interpreted in 2 ways. First, NOD alone (ie, normoxia during the day) may not have important health consequences, and thus there is no benefit to correction of NOD. An alternate explanation is that, although supplemental oxygen prevents NOD, any benefit is outweighed by side effects or harm from supplemental oxygen. Anecdotally, the administration of supplemental oxygen causes hypercapnic respiratory failure in those with lung disease. Supplemental oxygen might lead to hypercapnia via several mechanisms.121 First, administration of oxygen may reverse appropriate hypoxemic vasoconstriction and increase ventilation-perfusion mismatch. For example, patients with emphysema should appropriately shunt blood away from poorly ventilated lung units that are hypoxic. Oxygen administration will improve local hypoxia and blood will flow into these lung units, but the excretion of delivered CO2 will still be impaired. The Haldane effect, which describes the affinity of hemoglobin for carbon dioxide in the presence of oxygen, is also important. As PaO2 increases, the affinity of hemoglobin for CO2 decreases. Thus, as PaO2 rises, less CO2 will be bound to hemoglobin and more will be dissolved in plasma (increased PaCO2). It is the unbound level of CO2 that then crosses the blood-brain barrier and contributes to CO2 narcosis. Although frequently invoked, suppression of the hypoxemic respiratory drive, if any, is likely a minor effect. In those with lung disease, supplemental oxygen may have additional effects that allow for hypercapnia. Reversal of hypoxemia (perhaps after days of illness and dyspnea) may allow patients to sleep (leading to hypercapnia as discussed above). Sleep and the relief of dyspnea may increase the effective delivery of oxygen (by reducing inspiratory flow and entrainment of room air), which will perpetuate and exacerbate the effects of oxygen therapy and worsen hypercapnia.122 Only a few small studies have examined this issue. Samolski and colleagues measured the effect on morning PaO2 and PaCO2 of either baseline (daytime) supplemental oxygen or an additional 1 L/min (as per current guidelines) in those with hypercapnic COPD.112 The recommended increase in nocturnal supplemental oxygen was more effective at preventing NOD, but PaCO2 increased and pH decreased slightly after the single night of intervention. More prolonged treatment may contribute to more substantial hypercapnia. The detrimental effects of oxygen may be more pronounced in those who also have OSA. In a single study, Alford and colleagues administered 4 L/min supplemental oxygen to 20 men with both OSA and COPD. While nocturnal oxygenation improved, duration of obstructive events increased from 25.7 seconds to 31.4 seconds, resulting in an end-apneic PaCO2 increase from 52.8 to 62.3 mm Hg, with corresponding decreases in pH.123
Supplemental oxygen also has effects on cardiac function, not mediated via hypercapnia. An entire literature exists around the detrimental effects of supplemental oxygen in those with myocardial infarction or congestive heart failure.124 Acute administration of oxygen can reduce cardiac output, increase pulmonary capillary wedge pressure, and reduce coronary blood flow, even in those without heart disease.125,126 Coronary blood flow can be restored with administration of anti-oxidants, implying a causative role of oxidative stress in the oxygen-induced hemodynamic impairment.127 Such effects might be important in those with COPD, many of whom have heart disease and die most commonly of cardiovascular disease. Again, ongoing large trials will be helpful in this regard. Whatever the mechanism, the potential for harm suggests that, like any drug, supplemental oxygen use should be titrated only as high as needed to relieve hypoxemia during the night. By avoiding marked hyperoxia, detrimental effects of oxygen supplementation should be minor. After initiation of therapy, patients should be observed for development of hypercapnia, and these patients may benefit from noninvasive positive-pressure ventilation (see below).110 Hypercapnia could cause symptoms such as headache; however, an arterial blood gas sample will likely be needed to assess for an increase in PCO2.
Treatment of the Underlying Pulmonary Disease
If possible, treatment of the underlying lung disease is perhaps the best way to prevent or treat NOD. Again, most data come from patients with COPD, although we may be able to extrapolate these data to other (partially) reversible obstructive diseases such as asthma and cystic fibrosis. In moderate to severe COPD, ipratropium inhaled 4 times a day improved nocturnal oxygen saturation, and subjective sleep quality was better as well.128 Tiotropium and long-acting β agonists also improve nocturnal oxygen saturation.129,130 Oral steroid therapy in stable COPD has been shown to improve NOD and, surprisingly, increase total sleep time.131 Although there are no definitive data, we might expect a similar improvement with the use of inhaled corticosteroids. Taken together, the data suggest that treatment of underlying COPD will ameliorate NOD. In the NOTT study, a period of “stabilization” after enrollment but before randomization improved oxygenation so much that many subjects no longer qualified for long-term oxygen therapy. As above, many patients are placed on “long-term” oxygen therapy after an acute worsening of lung disease, and no longer qualify 1–3 months later.
Noninvasive Ventilation
One other way to augment nocturnal ventilation and possibly ameliorate NOD is with noninvasive ventilation (NIV), which has been trialed in stable hypercapnic COPD with multiple (small) studies and inconsistent results over the years.132 The largest and most recent trial, by McEvoy and colleagues, used NIV in patients with stable hypercapnic COPD already on long-term oxygen therapy, and showed a significant improvement in adjusted mortality.133 However, the improvement in mortality with NIV was associated with a worse quality of life in the NIV treat group. Windisch and colleagues use what they call “high-intensity NIV,” with very high driving pressures (average inspiratory pressure 28 cm H2O, average expiratory pressure 5 cm H2O) and a high breathing frequency of about 21 breaths/min in those with severe COPD. With these settings, which required in-hospital acclimatization, they see improvements in spirometry and blood gas abnormalities, including an improvement in daytime oxygen saturation.134 NIV may have a role in those with substantial sleep hypoventilation in response to supplemental oxygen therapy.
Summary
The physiologic changes that occur in ventilation with sleep contribute to NOD in those with lung disease. The optimal treatment of NOD would be improvement in the underlying lung disease, if possible. Nocturnal supplemental oxygen therapy should be used as part of continuous oxygen therapy in those with hypoxemia during sleep and wake. Ongoing trials may help to address whether nocturnal supplemental oxygen should be used in those with only desaturation during sleep. Until data are available, one approach would be an empiric trial of supplemental oxygen, with later evaluation to determine if there has been any subjective or objective improvement. If used, oxygen should be dosed as needed, and patients should be monitored for hypercapnia. Because of its prevalence, OSA may overlap with lung disease in many patients. If present, OSA should be treated. Patients with overlap syndromes may be good candidates for NIV.
Discussion
MacIntyre:
Bob, thank you very much. Let me ask you about oximetry at night, because it's a very challenging area. What if I do oximetry on somebody that I'm concerned about, and they're flat-out normal, or at least it doesn't drop below 90% for any appreciable time overnight? Does that effectively rule out important NOD issues?
Owens:
I think it definitely rules out NOD.
MacIntyre:
But are the oximeters good enough to catch short desaturation periods?
Owens:
To rephrase your question from my sleep perspective: Would nocturnal oximetry rule out somebody who had important sleep apnea? The answer is no. You can still have patients who have frequent arousals and all the effects of increased sympathetic tone, like elevated blood pressure and blood glucose. So I wouldn't use it for sleep apnea. If I was pretty sure it was just lung disease and I wanted to make sure that the patient didn't need supplemental O2 during the night, I think it would be good enough. I hear what you're saying. I think they're good for telling you what the O2 level is, and I think the time sensitivity is close enough for that purpose, but they don't tell you what's happening with upper airway flow, respiratory effort, and arousals.
McCoy:
You indicated to treat O2 like a drug. Have you tested O2-conserving devices on sleeping patients, and, if you haven't, do you have an opinion on that?
Owens:
I have not. I don't have an opinion. I would need to know more about it. But I would think that what you're getting at is the same thing I'm saying, which is try to use as little as you need. And on-demand therapy would be a fantastic thing.
Heffner:
I'm surprised that patients without OSA who have COPD and desaturate at night don't have some benefit from O2 therapy, considering the benefit of oxygen for daytime hypoxemia. Do you think part of our problem is that we haven't come up with a good diagnostic threshold of the degree and duration of hypoxemia to adequately identify the group that may benefit from O2 at night?
Owens:
That's certainly true. We've only gotten to the level of putting a bunch of people in a room and asking what everybody considers an important level and duration of desaturation, and I don't know that it's ever been correlated with, let's say, pulmonary artery pressure. Testing a working definition of what is a clinically important level of NOD would be very helpful. I was surprised as well, and certainly the early studies seemed to indicate, that O2 benefits sleep quality, and very modestly decreased pulmonary artery pressure in the Fletcher studies.1–4 I would go back to the combined data of the MRC trial5 and NOTT6: there isn't much separation until you get to continuous O2 therapy. Most of the other 3 arms are overlapping. It may also be that, if you give people O2 during the night via a stationary device, maybe they just sit in bed and are tethered to that bed for 10 hours a day instead of sleeping for 7 or 8 hours and getting up and walking.
Criner:
This follows up on Neil's question. It doesn't look like account has been made for the severity of the patients and the dynamic processes that happen in patients over time, due to the nature of their underlying disease, to know how often or when we need to check for NOD. We know those patients who have progressive disease have an increased number and severity of exacerbations. And we know that some patient groups have more frequent or severe exacerbator phenotypes. And we would think that their physiology of oxygen saturation that occurs at night is potentially compounded by changes in the instability of their disease, which would make their O2 need in response to NOD more profound. Do you think this is a gap in our knowledge: what patient groups and how homogenous or heterogeneous they are in their NOD profiles?
Owens:
Yes, the degree of NOD is quite variable amongst patients, and the physiological studies of NOD have been very small. It would be helpful to know the mechanisms that lead to profound desaturation in some people and not others. Maybe there are subgroups we can treat, but we don't know yet. The goal might be to use sleep as a natural stress test, and risk-stratify the patients, similar to what we do with coronary disease and those who undergo stress testing. It would be great to be able to say, “Let's study you at night, and if you desaturate we're going to treat you aggressively in some way to slow the progression of disease.” Unfortunately, we don't yet have evidence that this approach works.
Kevin Ward:
What role is there for capnography in diagnosis? I know capnography's limitations in terms of respiration, and when people recover: we see this with conscious sedation when we use it, so we can see an overshoot and hypercapnia after a period of apnea or hypoventilation. And what about metabolism during sleep? Do you have other tools, such as near-infrared spectroscopy, which lets you look at the venous end of things and get an O2 extraction ratio and understand whether these people vasodilate to compensate during the episodes?
Owens:
I'm glad you brought that up. We were talking about the benefits of pulse oximetry, but one of the unintended consequences is that we tend to focus on oxygenation and not on CO2. And if the capnography technology were more readily available or affordable, I think it would be great to know when patients are becoming hypercapnic when we give them O2. We'd give some O2 and measure both oxygenation and CO2. The devices are, I think, accurate enough, whether you do transcutaneous or end-tidal CO2 monitoring, but for home use the cost is prohibitive. Metabolic activity during sleep hasn't been very well studied, because it's technically hard to do. Interestingly, basal metabolic rate will usually go down during sleep, but if you have OSA, your metabolic rate will go up during sleep, because you're really working to breathe.7 Most people with OSA, when they come in they say, “I'm going to start wearing this mask and it's going to help me lose weight,” and actually most people gain weight when they go on CPAP, because the most exercise they've been getting has been from struggling to breathe at night, and you've relieved that.8
Lewarski:*
We regularly examine and analyze the Medicare oxygen claims data. In reviewing the trends in device claims by the different Healthcare Common Procedure Coding System [HCPCS] codes and referencing the current oxygen coverage policy, 2 things stand out. First, there appears to be a de facto standard for determining clinically relevant NOD, which is the Medicare medical necessity coverage criterion that requires 5 cumulative minutes of desaturation at or below 88% in a sleep period. Second, when we look at the claims data and separate the number of patients who just have a stationary O2 system versus those who have both a stationary and an ambulatory system, the stationary only group accounts for about 40%. That is, 40% of the total Medicare home O2 patients have only a stationary O2 system prescribed, suggesting nocturnal use only. While it's anecdotal and may or may not be influencing clinical practice and prescribing, many companies have been promoting overnight oximetry as the basis for evaluating early-stage COPD and NOD. So it appears by the trends in device use and prescribing that the actual clinical practice may be far ahead of the academics in this area, as is evidenced by the large portion of patients getting O2 in the United States for nocturnal use only.
Claure:
If you recommend titration of O2, what saturation would you consider too high and what saturation would you consider hypoxemia? Would you consider the effects of hypoxemia on the respiratory control of the patient, perhaps worsening the spells of hypoxemia?
Owens:
About 10 years ago Peter Calverley wrote an article saying titrate the saturation to 88-93%.9 It's a Goldilocks kind of solution: not too much and not too little. But it's never been rigorously studied. Based on control of breathing and studies of pulmonary arterial pressure, you're probably safe if the saturation is somewhere above 85% or 88%. At what level does hypercapnia occur? I think that's an individual response to some extent, but I would aim for something less than 95%. Everything I just said is very much data-free and speculation, but that's probably the most accepted practice.
MacIntyre:
A practice I see commonly used is to do a 6-minute walk test or other simple exercise test, and if the patient desaturates, the prescription is written, “Use O2 during exercise and sleep,” the assumption being there's some kind of link between the stress test of sleep and the stress test of walking down the hall. What's the literature supporting that link?
Owens:
That's probably a reasonable approach. In general, the desaturation that happens during sleep is even more profound than what occurs during exercise. In a study by Mulloy10 the NOD was on the order of 13% or 14% change in O2 saturation, but during exercise was only about 5%. So it can be much more profound. I think that if it makes sense to give O2 during exercise and sleep alone, that's a reasonable way to help find out who's going to desaturate at night. However, a recent study11 concluded that the approach of studying a patient during exercise was useful but not perfect in predicting O2 desaturation at night. For some, you'd still have to do an overnight oximetry test, and you need to follow up with another test at night to see if you've adequately titrated the O2.
Branson:
Is there a need in the market for a noninvasive ventilator with a concentrator built into it, so we can give the benefits of both NIV and O2?
Owens:
Right now you can bleed in the O2, so I don't know the advantages of integrating them versus having the 2 systems we have now. Anecdotally, I think there are patients who may not need as much O2 or even don't need O2 at all if you completely take out the upper airway and increase their ventilation a little bit. An NIV device that measured oxygenation and CO2 would be the most helpful.
Branson:
Work we did recently was on closed-loop control of O2 in the ICU. Trauma and surgical patients tend to be hyperoxic: once the FIO2 is below 0.5, if the patient's PO2 is 160 mm Hg, nobody cares.
MacIntyre:
I care.
Branson:
My observation of these patients from the bedside is that when they are hyperoxic and go on closed-loop control and become normoxic, they're easier to arouse and more responsive to stimuli, and I'm wondering if there's a physiologic mechanism for that? I think it relates to pain control as well, but I don't know if that works with your descriptions of the physiology of O2.
Owens:
The concept of O2 toxicity at very high FIO2 is interesting. There are a lot of animal data but no human data as of yet. To get to your point about seeing patients in the ICU who may become more alert when you turn down the FIO2, I think there are 2 possible mechanisms.
The first, and I think the best described, is the Haldane effect. Hemoglobin can bind either CO2 or O2, but it prefers O2, so, for the same volume of blood, with a higher PaO2 you're going to have more CO2 dissolved in the blood: a higher PaCO2. Because of the blood-brain barrier, only PaCO2 (not CO2 bound to hemoglobin) can cause pH changes in the cerebrospinal fluid. If you give somebody a bunch of O2 and increase the PaO2, you can raise their PaCO2 5 to 10 mm Hg, and it's a very quick effect.
The second thing is that, in my experience, if a patient is on less O2, the staff are more comfortable about giving narcotics and getting pain under control. People say, “Oh, they're on 100% face-mask: we don't want to give too much pain medication.” So perhaps when the O2 is turned down, providers are more comfortable giving adequate amounts of analgesia, so the patient can interact more.
Branson:
My last question is related to the O2 in patients at risk for hypercapnic respiratory failure: not just COPD, but neuromuscular disease, morbid obesity, or obesity hypoventilation syndrome. The residents and respiratory therapists at our place would say that you don't give high-flow oxygen to patients with chronic lung disease because it “knocks out the hypoxic drive.” Nobody discusses the Haldane effect or really understands that phenomena, and nobody looks at hypoxic pulmonary vasoconstriction and how that changes ventilation perfusion. I think that's very important to understand.
Owens:
I completely agree. If an intern tells me something about V̇/Q̇ [ventilation-perfusion] mismatch—which is the answer to all pulmonary questions—that's a gold star for the day if they don't come out of the gate with hypoxemic respiratory drive as the cause for hypercapnic respiratory failure. There are 3 things to mention: V̇/Q̇ mismatch from release of appropriate hypoxic vasoconstriction; the Haldane effect; and if there is a hypoxemic respiratory drive—about which there is controversy—then it is likely a minor effect.
There are also a couple other things to consider. First, if you have somebody who hasn't been sleeping and they are sleep-deprived and now you give them O2, relieve dyspnea, and the patient goes to sleep, their CO2 will rise just from going from wake to sleep. Once asleep, minute ventilation falls, so the effective amount of supplemental oxygen will increase, which will worsen the hypercapnia via the above mechanisms. And there are other reflexes that might be important in decreasing dyspnea and minute ventilation with supplemental oxygen applied.
Pierson:†
A point implied by both Joe and Rich touches on a theme that has been recurrent through many of these conferences over the years, which is the distinction between what we do and what we know. They're both important, and one would hope that they would coincide completely, but we all know they don't, and one of the functions of these Journal conferences is to point out the distinctions and to try to bring them closer together.
Footnotes
- Correspondence: Robert L Owens MD, Division of Sleep Medicine, Brigham and Women's Hospital, 75 Francis Street, Boston MA 02115. E-mail: rowens{at}partners.org.
Dr Owens presented a version of this paper at the 50th Respiratory Care Journal Conference, “Oxygen,” held April 13–14, 2012, in San Francisco, California.
The author has disclosed no conflicts of interest.
↵* Joseph S Lewarski RRT FAARC, Invacare, Elyria, Ohio.
↵† David J Pierson MD FAARC, Emeritus, Division of Pulmonary and Critical Care Medicine, Harborview Medical Center, University of Washington, Seattle, Washington.
- Copyright © 2013 by Daedalus Enterprises Inc.
References
- 1.↵
- 2.
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.
- 68.
- 69.↵
- 70.
- 71.
- 72.
- 73.↵
- 74.↵
- 75.↵
- 76.
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 113.↵
- 114.↵
- 115.↵
- 116.↵
- 117.↵
- 118.↵
- 119.↵
- 120.↵
- 121.↵
- 122.↵
- 123.↵
- 124.↵
- 125.↵
- 126.↵
- 127.↵
- 128.↵
- 129.↵
- 130.↵
- 131.↵
- 132.↵
- 133.↵
- 134.↵









{kind=link}
{kind=link}
{kind=link}
{kind=link}