

Abstract
Pulmonary arterial hypertension (PAH) is a disease of small pulmonary arteries, characterized by vascular proliferation and remodeling. Progressive increase in pulmonary vascular resistance ultimately leads to right ventricular heart failure and death. PAH-specific drug therapy has improved clinical outcomes and survival. While the survival is better, progression of pulmonary vasculopathy contributes to pulmonary artery dilatation. Left main compression syndrome, pulmonary artery dissection, pulmonary artery rupture, and severe hemoptysis are reported as complications leading to sudden cardiac death, an event encountered more often in PAH patients. The advent of PAH-targeted drug therapy has reduced referral for lung transplantation; however, severe complications require rapid diagnosis, decision making, and possible registration on a lung transplantation waiting list. PAH referral centers provide multidisciplinary emergency care and specific therapeutic management, contributing to improved quality of life and survival for PAH patients. We review the complications leading to sudden death in PAH.
- pulmonary hypertension
- pulmonary artery dissection
- pulmonary artery rupture
- left main compression syndrome
Introduction
Pulmonary arterial hypertension (PAH) is a devastating disease, leading to right ventricular (RV) heart failure and death. Two decades ago, the median survival rate from diagnosis, despite the available supportive treatment,1 was less than 3 years. In the current era, 8 drugs from 3 pharmacologic classes (endothelin receptor antagonists, phosphodiesterase-5 inhibitors, and prostanoids), administered per os, via inhalation, subcutaneously, or intravenously, have been approved for PAH patients, improving survival. While the survival is better, pulmonary hypertension continues to cause substantial morbidity and mortality, as progression of the pulmonary vasculopathy leads to progressive RV failure.2 Moreover, new imaging modalities enable us to recognize major complications, previously missed.
Sudden cardiac death is now encountered more often in PAH patients. In the American National Institute of Health registry, 106 deaths were reported in a cohort of 194 patients with idiopathic PAH, of which 26% were sudden.3 Likewise, 99 out of 316 patients died in the Leuven database during follow-up, of whom 18 suddenly.3
We review the etiology, preventive measures, and management of complications associated with sudden cardiac death among PAH patients.
Plausible Causes of Arrhythmia
The mechanisms of sudden cardiac death associated with RV hypertension and arrhythmias are less well understood than are those associated with left ventricular (LV) disease. Arrhythmogenic substrate in rat hearts with monocrotaline-induced pulmonary hypertension may cause steeper RV electrical restitution and rate-dependent RV-LV action potential duration-dispersion,4 inducing ventricular tachycardia and fibrillation. A study of 201 pulmonary hypertension patients5 demonstrated that mean heart-rate corrected QT interval (QTc) and QTc dispersion (QTcd) were significantly increased in severely ill patients, compared to patients with mild to moderate pulmonary hypertension. In addition, in women these indices were positively correlated to mean pulmonary arterial pressure, identifying a possible substrate for ventricular arrhythmias.
Ventricular arrhythmias in PAH are predominantly described in patients with congenital heart disease. Congenital heart disease patients at increased risk for sudden cardiac death are those with tetralogy of Fallot, transposition of great arteries, congenitally corrected transposition of the great arteries, aortic stenosis, and univentricular heart disease.6,7 In these patients, arrhythmias represent an increasingly frequent cause of morbidity and mortality,8,9 but algorithms for risk stratification have not yet been established.9 Implantable cardioverter defibrillator is indicated for secondary prevention, provided that a reversible cause for the cardiac arrest has been excluded. Electrophysiologic study is indicated for spontaneous sustained ventricular tachycardia, which can be eliminated by catheter ablation or surgical resection.9 In patients with Eisenmenger physiology, supraventricular arrhythmias may predispose to sudden cardiac death, and it is therefore essential to restore and maintain sinus rhythm.10 In a series by Daliento et al, 42% of Eisenmenger patients were found to have supraventricular arrhythmias on routine electrocardiogram or 24-hour Holter monitoring during long-term follow-up.11
In contrast to patients with pulmonary hypertension due to left heart disease, malignant ventricular arrhythmias, such as ventricular tachycardia or ventricular fibrillation, are rarely present in PAH patients.12 In a series of 132 PAH patients with cardiac arrest, by Hoeper et al,13 ventricular fibrillation was found in only 8% of the cases. The hypothesis that pulmonary hypertension promotes spontaneous ventricular fibrillation in rats, during a critical post-pulmonary-hypertension onset period, was tested in a recently published study.14 The authors concluded that pulmonary-hypertension-induced RV fibrillation is associated with a distinct phase of increased mortality, characterized by spontaneous ventricular fibrillation arising from the RV by an early after-depolarization-mediated triggered activity.
In contrast, supraventricular tachyarrhythmias are quite frequent. In a study15 of 231 PAH patients followed up for 6 years, supraventricular arrhythmias were reported with an annual incidence of 2.8%, whereas the incidence of atrial flutter and of fibrillation were almost equal, leading to rapid clinical deterioration. Atrial flutter, originating from the right atrium, seems to occur more often in patients with severely impaired hemodynamics, but does not seem to be the substrate for sudden cardiac death.16
Atrial tachyarrhythmias are poorly tolerated in PAH because of decreased ventricular compliance, which renders RV filling dependent on the atrial contraction.17 Clinical improvement in PAH patients after atrial flutter isthmus ablation has been described.16
Non-arrhythmic Causes
The most relevant mechanisms for sudden cardiac death in PAH patients seem to be related to severe dilatation of the pulmonary artery, as subsequent complications, such as left main compression syndrome (LMCS), pulmonary artery dissection (PAD), pulmonary artery rupture (PAR), and massive hemoptysis, may take place.
Left Main Compression Syndrome
Pulmonary artery dilatation represents an important consequence of PAH, and is commonly seen in echocardiographic studies as well as on computed tomography. Pulmonary artery dilatation is progressive and, surprisingly, is independent of the changes in pulmonary artery pressure, cardiac output, and even hemodynamics.18
LMCS due to extrinsic compression of the left main coronary artery by an enlarged pulmonary artery trunk is an uncommon cause of angina, LV dysfunction, and sudden cardiac death in patients with pulmonary hypertension19 (Fig. 1). According to various case studies,20–22 the true incidence of LMCS due to pulmonary artery dilatation is unknown but ranges from 5% to 44% of patients with pulmonary hypertension. A prominent risk factor for the development of LMCS seems to be the severity and duration of pulmonary hypertension.

Dual source computed tomogram used for pulmonary artery diameter measurement and left main compression syndrome detection.
Angina in PAH patients is a frequently reported symptom, predominantly caused by RV subendocardial ischemia due to RV dilatation and hypertrophy; however, cases of acute coronary syndrome23–25 or LV failure and cardiogenic shock24–26 have been reported. Ventricular tachyarrhythmias due to ischemia secondary to LMCS might contribute to an increased risk of sudden cardiac death in these patients. So, in case of angina in a patient with pulmonary hypertension, LMCS should be considered in the differential diagnosis.
The syndrome has been described in the setting of congenital heart defects such as atrial27 and ventricular septal defects and patent ductus arteriosus.28 Other underlying etiologies are idiopathic PAH, chronic thromboembolic pulmonary hypertension, and advanced parenchymal lung disease.28–30 Several structural changes are responsible for the extrinsic left main coronary artery compression, and pulmonary artery vascular remodeling seems to be crucial. In case of chronic pulmonary hypertension, intimal thickening, medial hypertrophy, fibrosis, and luminal dilatation appear in proximal pulmonary vessels. Pulmonary artery dilatation may lead to the displacement of the left main coronary artery. In a series28 by Kajita et al, pulmonary artery dilatation was present in all cases, with a mean main pulmonary artery-to-aortic root diameter ratio of 2.0. This was also confirmed by Mesquita et al,20 who reported, in a series of patients with pulmonary hypertension, a mean pulmonary artery diameter of 55 mm and a mean pulmonary artery-to-aortic root diameter ratio of 1.98 in patients with LMCS, compared with 37 mm and 1.46 in those without.
Diagnosis of Left Main Compression Syndrome.
In the presence of substantial dilatation of the main pulmonary artery, further evaluation should be performed to exclude LMCS,25 especially in patients with angina, as the likelihood of left main coronary artery compression in patients with PAH is positively related to both pulmonary artery diameter and the ratio of pulmonary artery diameter to aortic diameter.28 Cardiac computed tomography or magnetic resonance angiography are useful tools for noninvasive screening31; coronary angiography,25 however, is considered the gold standard for the final diagnosis of LMCS.32
Left main coronary artery compression is usually best visualized in the 45° left anterior oblique view, with 30° cranial angulation.25,33 In this projection the left main coronary artery has an eccentric narrowing and appears to be inferiorly displaced, in close contact with the left aortic sinus,25 with a mean angle of 23°, compared with 70° in the control group. Intravascular ultrasound study and fractional flow reserve estimation have also been used to evaluate the compression severity.23,32,34 Slight narrowing of the ostial left main coronary artery, without evidence of substantial atherosclerosis, is always present. Myocardial perfusion techniques do not seem to be of any help in establishing the diagnosis; according to the reported cases in the literature, only 4 out of 10 patients with documented LMCS had evidence of regional ischemia on nuclear myocardial imaging.28,33,35
Management of Left Main Compression Syndrome.
In case of LMCS it is crucial to restore unobstructed coronary flow; this seems to reduce the incidence of sudden cardiac death. Treatment is indicated when angiographic compression is documented; noninvasive evaluation of myocardial ischemia does not seem to be of interest in this setting.32
The optimal therapeutic approach is, however, debatable. Surgical correction of the dilated pulmonary artery has been reported,25 and is associated with a reduction in left main coronary artery stenosis, from 85% to smaller than 50%, as well as less inferior left main displacement. Coronary revascularization, however, is the surgical procedure of choice.36
Percutaneous coronary intervention combined with stent implantation seems to be a safe and effective option, avoiding the postoperative risk of RV failure in patients with increased pulmonary arterial pressure.31 Due to the absence of atherosclerotic disease, the risk of percutaneous intervention in these patients seems to be low. The lesion is most often ostial, and stenting is feasible, with a high procedural success rate and a low restenosis risk at follow-up. In 2001, Rich et al37 reported successful left main coronary artery stenting in 2 patients with primary pulmonary hypertension and LMCS. Since then, several other authors23,38–41 have also reported successful angiographic and short-term clinical outcomes. Of note, all reported cases involved compression of the ostium or proximal left main coronary artery, sparing the left main bifurcation, so single stent placement was always sufficient.
Pulmonary Artery Dissection and Rupture
In patients without pulmonary hypertension, rare cases of idiopathic and inflammation-related PAD have been described.42,43 PAD and PAR have also been proposed as the underlying pathology in PAH patients presenting with cardiogenic shock and sudden death,44 more frequently diagnosed in Eisenmenger syndrome.45–49 PAD is related to medial degeneration, with fragmentation of elastic fibers, weakening of the wall, and dilatation of the pulmonary artery and its branches caused by chronic pulmonary hypertension.45,50,51 The increased intravascular pressure and subsequent shear stress may predispose to the development of an intimal tear. Whether medial degeneration causes the dissection, predisposes to intimal tears, or is simply the result of chronically elevated intravascular pressure remains controversial.47
Approximately 70 cases of PAD have been described,43–46,50–69 of which almost 10 were diagnosed during life.43,65–69 Congenital heart disease was the underlying condition in the majority of cases, with the patent ductus arteriosus representing the most common defect. Idiopathic PAH was present in 10 cases.
The main pulmonary artery trunk is the site of dissection in 80% of cases. In a small number of cases, PAD may occur at the site of localized aneurysms, which are most common in congenital heart disease.67 In contrast to aortic dissection, the false lumen in PAD tend to rupture rather than to develop a re-entry site.68 PAR may occur into the pericardium70–72 or pleural cavity, leading to sudden death, and usually involves the site of maximal diameter of the pulmonary artery.
The precipitating factor of death in 20 among 182 pulmonary hypertension patients was pulmonary artery dilatation, according to a recent study in Poland. Multivariate analysis identified pulmonary artery diameter (P < .001) as independently contributing to the risk of sudden death.73
Iatrogenic (catheter-induced) rupture of the pulmonary artery is also a rare and life-threatening complication of right heart catheterization, and demands rapid therapy. Diffuse pulmonary bleeding or hemoptysis during right heart catheterization should immediately raise suspicion of iatrogenic PAD.
Diagnosis of Pulmonary Artery Dissection and Rupture.
The diagnosis of PAD and PAR is usually made postmortem, as the majority of these patients experience sudden death.70 High suspicion is needed in a PAH patient presenting with acute dyspnea on exertion, retrosternal chest pain, central cyanosis, and sudden hemodynamic decompensation.69 Symptom initiation may occur during exercise, as an acute increase in pulmonary arterial pressure, combined with the inflammatory substrate in PAH.
The echocardiogram, because of its accessibility, remains a first line diagnostic tool, but contrast-enhanced computed tomography pulmonary angiography, or magnetic resonance angiography,74 represents a powerful imaging modality in PAD and PAR (Fig. 2) for a timely surgical repair.
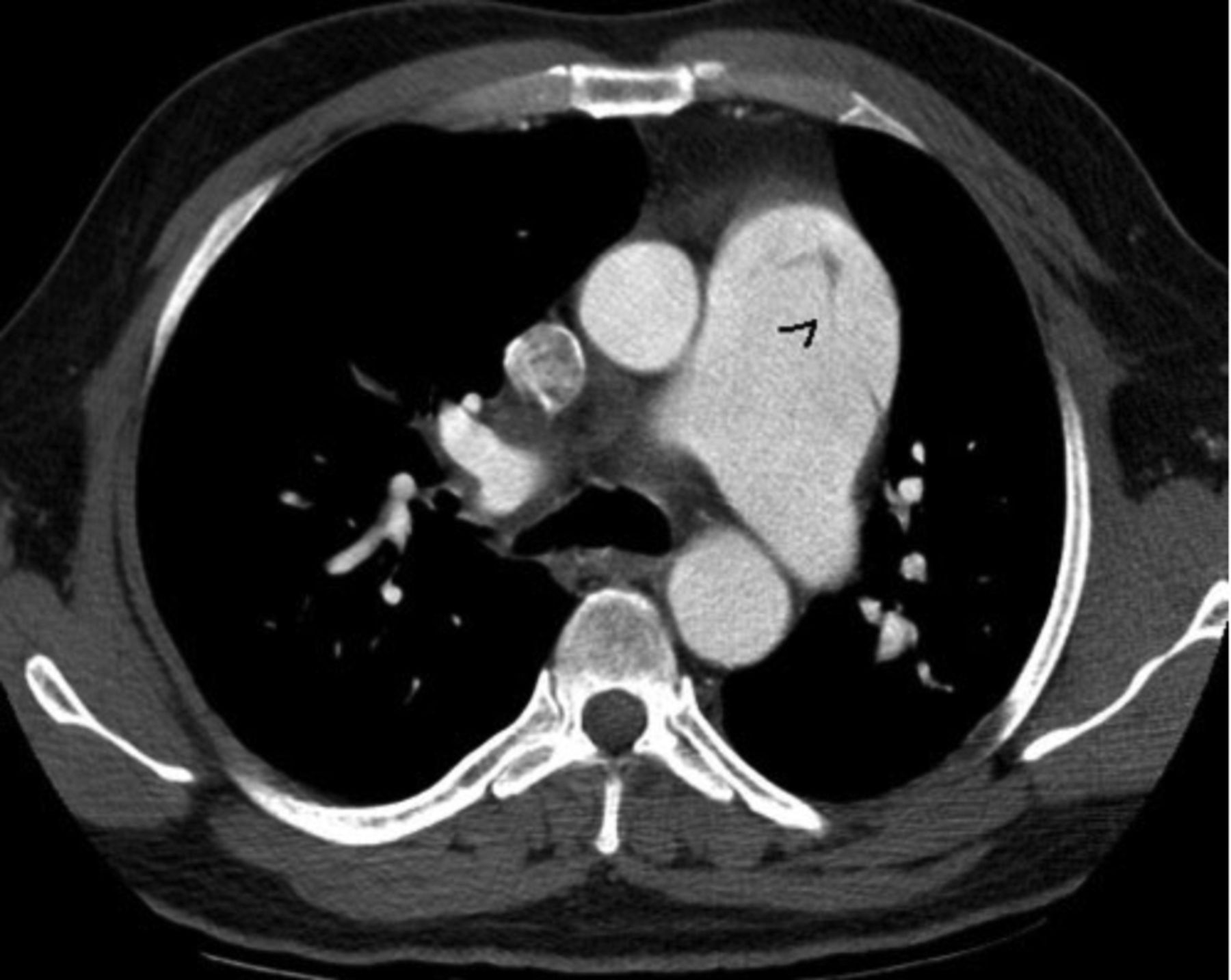
Contrast-enhanced computed tomography pulmonary angiogram showing dilated central pulmonary arteries and an intimal flap in the main pulmonary artery (black arrowhead). (From Reference 74, with permission.)
Management of Pulmonary Artery Dissection and Rupture.
The low likelihood of this complication in PAH patients has not allowed for widely accepted management guidelines. The majority of all PAD cases described in the literature are iatrogenic, and various procedures have been performed. They include lung isolation in patients requiring intubation to protect the contralateral lung and to decrease bleeding in the affected lung,75 endovascular techniques such as metal-coil embolization,76 stent graft implantation leading to successful sealing of the pulmonary perforation,77 therapeutic embolism78 of the segmental artery by using a liquid, tissue-adhesive, occlusive agent (isobutyl-2-cyanoacrylate), embolization of the vessel by injection of thrombin,79 and embolization with gelatin foam.80
PAD is not often be encountered in PAH patients,48,81 but besides the above treatment options, urgent heart-lung transplantation has been reported in experienced centers,47–49,82 and seems to be the necessary approach.
Hemoptysis
Massive hemoptysis is one of the most dreaded respiratory emergencies and is caused by various underlying mechanisms.83 Severe hemoptysis leading to uncontrolled bleeding and sudden death appears to be uncommon, with a mortality rate exceeding 50% if appropriate treatment is not immediately provided.84–88
The source of massive hemoptysis89 is predominantly the bronchial circulation (90%), rather than the pulmonary circulation (5%), and in a minority of cases it may originate from the aorta or the systemic arterial supply to the lungs.90–92 In PAH the hypoxic vasoconstriction and the intravascular thrombosis93 reduce pulmonary circulation, resulting in bronchial artery proliferation and enlargement.89,94,95
The prominent collateral vessels and the hypertrophy of bronchial collateral arteries correlate with disease severity96 and sometimes induce extravasation into the respiratory tract, resulting in massive hemoptysis.97
According to the French national reference center experience, in contrast to PAH associated with congenital heart disease, idiopathic and heritable PAH hemoptysis represents a rare complication, with 20 cases reported over a 10-year period.98 In chronic thromboembolic pulmonary hypertension, hemoptysis occurs more often and may be recurrent, as a result of dilated and hypertrophied bronchial collateral circulation.99 This complication can be life-threatening, with cumulative blood volumes averaging 79 mL (20–300 mL). Survival rates of 60%, 43%, and 36% at 1, 3, and 12 months, respectively, were documented, according to the French national reference center experience.98
Since many PAH patients are treated with anticoagulants, therapeutic dilemma may ensue in case of concomitant hemoptysis.
Diagnosis of Hemoptysis.
Diagnostic options for massive hemoptysis include radiography, bronchoscopy, and computed tomography, in an effort to elucidate the underlying cause as well as the exact location of the bleeding.100–102 Routine chest x-ray is readily available and helpful; however, in a retrospective evaluation of 208 patients with hemoptysis, Hirshberg et al103 found that radiography was diagnostic in only 50% of cases. Bronchoscopy is by far more accurate, but the role of fiberoptic bronchoscopy in the setting of massive active hemoptysis is still controversial. The excessive blood in the bronchi, the risk of airway compromise by sedation, the delay in definitive treatment, the hypoxia, and the high cost are the main drawbacks of bronchoscopy. Computed tomography is extremely valuable in localization of bleeding, with higher accuracy than bronchoscopy,100,104 since it can detect both bronchial and non-bronchial vessels.
Management of Hemoptysis.
Treatment of hemoptysis in PAH patients is not different from other causes of hemoptysis, for which there is no management consensus. Neutralization of oral anticoagulants with vitamin K, and reversal of heparin with protamine, administration of the antifibrinolytic tranexamic acid, bronchoscopy, airway protection with balloon tamponade or double-lumen endotracheal tube, and selective embolization are the usual steps.3
Since the bronchial circulation is the major source of hemoptysis, in selected patients therapeutic embolization can be life-saving.105 In order to embolize the responsible vessel, a detailed angiography of the bronchial and pulmonary vascular tree is required. The concomitant embolization of non-bronchial systemic arteries at the same setting is favored if they are angiographically shown to contribute to the blood supply. Embolization of bronchial collaterals has been proposed in order to avoid recurrences of hemoptysis in patients with PAH. The selection of arteries to be embolized is based on the findings of computed tomography, bronchoscopy, and angiography, always in relation to the clinical situation.106 It has been proposed that repeated embolizations should not be considered as a definitive treatment in patients with PAH with recurrent bleeding.107
The complications of embolization have diminished gradually over the years, and include subintimal dissection of a bronchial artery, bronchial arterial perforation by a guide wire, and the reflux of embolic material into the aorta.105 Chest pain is the most common complication, possibly related to ischemia; dysphagia has also been reported.108,109 The most disastrous complication110,111 is spinal cord ischemia due to the inadvertent occlusion of spinal arteries, with a prevalence of 1.4–6.5%.
Syncope in PAH
Syncope is characterized by transient loss of consciousness due to cerebral hypoperfusion, with rapid onset, short duration, and spontaneous complete recovery. It occurs from low cardiac output and represents a grim prognostic sign in PAH patients, requiring immediate treatment.112 The incidence of syncope in newly diagnosed adult patients in the current era is 12%.113 Syncope increases the risk of death, and this is incremental to the risk attributable to other known prognostic factors.
Echocardiographic assessment provides useful information about pulmonary hemodynamics, but right heart catheterization is necessary to establish the diagnosis. Inotropic drugs and intravenous prostanoids are indicated to clinically stabilize the patient. Few studies have addressed the value of vasopressors and pulmonary vasodilators in critically ill PAH patients, as well as in patients successfully resuscitated after sudden cardiac death, but dobutamine, milrinone, inhaled nitric oxide, and intravenous prostacyclin are commonly utilized.114
Cardiac Arrest and Resuscitation in PAH Patients
Cases of sudden cardiac death due to previously undiagnosed PAH have been described. The diagnosis in these cases is based on autopsy, and on the pathophysiological changes that apparently exist in PAH such as RV myocardial hypertrophy, dilated pulmonary conus, plexiform vascular lesions, and thrombotic lesions. In case of successful resuscitation, echocardiographic assessment and right heart catheterization are necessary for PAH diagnosis establishment.
Cardiopulmonary resuscitation in PAH patients has poor outcome, as shown in the retrospective survey by Hoeper et al.13 In a population of 3,130 PAH patients treated between 1997 and 2000 in 17 reference centers in Europe and the United States, 513 patients had a circulatory arrest. Resuscitation was unsuccessful in 79% of patients (104 patients), and only 6% (8 patients) survived for longer than 3 months.
According to a recent review, cardiopulmonary resuscitation is not indicated in patients with a combination of: New York Heart Association class IV symptoms; intractable right heart failure with more than 2 hospital admissions over the preceding 6 months; maximal PAH specific drug therapy (including parenteral prostaglandin I2); atrial septostomy if indicated; contraindication for lung transplantation; and persistent intolerable suffering from dyspnea, anxiety, and pain.3
Summary
PAH is a rare and severe disease characterized by pulmonary vascular remodeling, leading to right heart failure and premature death. LMCS must be taken into consideration in PAH patients with angina, as well as in those without symptoms but with high-risk anatomy, as in case of severe pulmonary artery dilatation. Computed tomography coronary angiography represents the initial method for LMCS exclusion. Coronary angiography should be performed when the findings of computed tomography coronary angiography are suspicious. Coronary revascularization is of vital importance in these patients; in the current era, percutaneous revascularization with stent implantation seems to be safe and effective.
Massive hemoptysis, mostly due to PAR, is usually lethal in PAH patients with severely dilated pulmonary artery, so PAH patients with recurrent hemoptysis might be placed on the lung transplant list.
PAD is rarely described in surviving patients; it represents a life-threatening condition that should be suspected in PAH patients presenting with chest pain or hemodynamic compromise. Nowadays, the high quality noninvasive imaging techniques allow us to diagnose this condition and subsequently treat it surgically.
PAH may present with various complications that may cause sudden cardiac death; appropriate diagnostic approach, rapid decision making, and successful management should be applied. The advent of disease-targeted therapy for severe PAH has reduced patient referral for lung transplantation programs; however, the identification of severe complications such as PAD or recurrent massive hemoptysis, once recognized, are indications for rapid registration on a waiting list.
Finally, it is now appreciated that specialized multidisciplinary teams in PAH referral centers provide emergency care, direct links, and quick referral for lung transplantation or thoracic surgery, thus contributing to improved quality of life and increased survival for PAH patients.
Footnotes
- Correspondence: Eftychia A Demerouti MD PhD MmedSc, Cardiology Department, Onassis Cardiac Surgery Center, Syngrou Avenue 356, Athens, 17674, Greece. E-mail: efidemer{at}otenet.gr.
The authors have disclosed no conflicts of interest.
- Copyright © 2013 by Daedalus Enterprises
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.
- 40.
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.
- 53.
- 54.
- 55.
- 56.
- 57.
- 58.
- 59.
- 60.
- 61.
- 62.
- 63.
- 64.
- 65.↵
- 66.
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.
- 86.
- 87.
- 88.↵
- 89.↵
- 90.↵
- 91.
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.
- 102.↵
- 103.↵
- 104.↵
- 105.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 113.↵
- 114.↵





{kind=link}
{kind=link}