

Abstract
Hypercapnia and hypocapnia commonly complicate conditions that are present in critically ill patients. Both conditions have important physiologic effects that may impact the clinical management of these patients. For instance, hypercapnia results in bronchodilation and enhanced hypoxic vasoconstriction, leading to improved ventilation/perfusion matching. Hypocapnia reduces cerebral blood volume through arterial vasoconstriction. These effects have also been exploited for therapeutic aims. In patients with traumatic brain injury (TBI), hypocapnia is often utilized to control intracranial pressure. However, this effect is not sustained, and prolonged hypocapnia increases the risk of mortality and severe disability in patients with TBI. Hypercapnia and hypercapnic acidosis are common consequences of lung-protective ventilation in ARDS. Hypercapnic acidosis reduces ischemic lung injury and preserves lung compliance, but concern has arisen over hypercapnia-induced immunosuppression and the potential for bacterial proliferation in sepsis. Experimental studies suggest that buffering hypercapnic acidosis attenuates these effects, whereas hypocapnia appears to potentiate lung injury through increased capillary permeability and decreased lung compliance. Several areas of uncertainty surround the role of hypercapnia/hypocapnia in treating TBI and ARDS. Current data support recommendations to avoid hypocapnia in treating TBI, with the exception of emergent treatment of elevated intracranial pressure, while awaiting definitive management. Permissive hypercapnia is commonly accepted as a consequence of lung-protective ventilation in ARDS, but there is insufficient evidence to support the induction of hypercapnic acidosis in clinical practice. Buffering hypercapnic acidosis should be considered only for a specific clinical indication (eg, hemodynamic instability). For clinicians choosing to buffer hypercapnic acidosis, tris-hydroxymethyl aminomethane is recommended over sodium bicarbonate, as it is more effective in correcting pH and is not associated with increased carbon dioxide production. Future studies should aim to address these areas of uncertainty to help guide clinicians in the therapeutic use and management of hypercapnia/hypocapnia in critically ill patients.
- hypercapnia
- hypocapnia
- intensive care units
- respiration, artificial
- respiratory acidosis
- respiratory distress syndrome, adult
Introduction
In the ICU, hypercapnia and hypocapnia are common complications of conditions in critically ill patients. Although arterial CO2 levels (PaCO2) represent the amount of CO2 dissolved in blood, carbon dioxide is also present in bicarbonate ions and bound to hemoglobin. In contrast, the majority of oxygen carried in blood is bound to hemoglobin, whereas very little is dissolved. PaCO2 is impacted by alveolar ventilation, CO2 production, and the fraction of inspired CO2.1
One major pulmonary function is to continually transport air from the atmosphere to the gas exchange units of the lung, including the alveoli, alveolar sacs, alveolar ducts, and respiratory bronchioles. The rate at which new atmospheric air reaches the gas exchange units of the lung refers to the rate of alveolar ventilation. This is influenced by the breathing frequency (f) and the dead space (VD) occupying each tidal breath (Equation 1, where VT is tidal volume).2
 (1)
(1)
The total VD is composed of the anatomic and physiologic VD. Air that fills a conducting airway but does not usually participate in gas exchange during a tidal breath is said to occupy the anatomic VD. This is composed of the nasopharynx, oropharynx, trachea, bronchi, and bronchioles. When alveoli are rendered nonfunctional (eg, in COPD) or do not receive adequate perfusion to participate in gas exchange, they contribute to the physiologic VD. The rate of alveolar ventilation has a major impact on systemic CO2 and oxygen levels.2,3 Most commonly, PaCO2 levels are altered in response to changes in alveolar ventilation, where respiratory depression (ie, due to sedatives or narcotics),4 airway obstruction,5 and increased VD can drive PaCO2 higher.6
Although the rate of carbon dioxide production generally remains constant, hypermetabolic states such as sepsis, malignant hyperthermia, thyroid crisis, and overfeeding can lead to elevations in CO2 levels (Table 1). The presence of fever and its associated hypermetabolic state have been noted to significantly impact PaCO2 levels, with each degree Celsius increase in core body temperature increasing CO2 production by 14%.7 When sodium bicarbonate is used in the clinical setting (eg, to buffer acidosis), it can also result in the generation of CO2. Finally, citrate-containing anticoagulants are commonly used in patients requiring dialysis. Citrate is metabolized in the liver to bicarbonate, which combines with hydrogen ions produced by organic acids and dissociates to produce CO2. In these patients, citrate may contribute to elevated PaCO2 levels. Finally, although the fraction of inspired CO2 is negligible in nonventilated patients, critically ill patients who are connected to a ventilator can become hypercapnic through rebreathing CO2 that accumulates in the breathing circuit.8
Common Causes of Hypercapnia and Hypocapnia
Although healthy individuals regulate blood CO2 levels through negative feedback mechanisms modulated by central and peripheral chemoreceptors,9,10 hypercapnia/hypocapnia may be intentionally or unintentionally induced in patients requiring mechanical ventilation. Importantly, it is now being recognized that hypercapnia and hypocapnia have physiologic implications that may affect patient-important outcomes.
In this review, we describe the physiologic impact of hypercapnia/hypocapnia and discuss their application in 2 distinct clinical settings: traumatic brain injury (TBI) and ARDS.
The Physiologic Effects of Carbon Dioxide
The Respiratory System
Carbon dioxide exerts its effect on airway resistance through influencing airway tone. Although hypercapnia is associated with bronchodilation,11 hypocapnia induces the opposite effect. Moreover, hypercapnia has been demonstrated to improve lung compliance through surfactant-independent mechanisms.12 Furthermore, hypercapnia increases pulmonary vascular resistance through enhancement of hypoxic vasoconstriction.13 In combination, these factors improve ventilation/perfusion matching and may facilitate improved gas exchange. Through inducing bronchoconstriction and inhibiting hypoxic vasoconstriction, hypocapnia inhibits gas exchange and increases intrapulmonary shunting.14
The Central Nervous System
Adjustments in PaCO2 levels and the associated changes in pH alter cerebral blood flow through impacting arterial vascular tone. Hypercapnia increases cerebral blood flow by 1–2 mL/100 g/min for every 1 mm Hg increase in PaCO2.15 Conversely, reducing PaCO2 to 20–25 mm Hg decreases cerebral blood flow by 40–50% via cerebral vasoconstriction.16
Additionally, hypocapnia increases neuronal excitability17 and cerebral glucose utilization.18 Due to the respiratory alkalosis that accompanies hypocapnia, the oxyhemoglobin dissociation curve is shifted to the left, reducing oxygen delivery to the brain. The imbalance between oxygen supply and demand in hypocapnia ultimately increases the risk of inducing cerebral ischemia.
Although hypocapnia has been induced in the clinical setting to reduce cerebral blood flow, it should be noted that its impact is limited to arterial blood flow, which accounts for a small percentage of overall cerebral blood volume.19 Finally, the effect of hypocapnia on cerebral blood flow is transient and largely reversed within hours due to metabolic compensation by the kidneys for reduced levels of PaCO2, resulting in reduced extracellular levels of bicarbonate. Because cerebral arterial blood flow is altered in a pH-dependent manner, the restoration of normocapnia after a period of sustained hypocapnia can result in normocapnic acidosis due to the relative bicarbonate deficiency, where the kidneys have not had time to compensate. Accordingly, the restoration of normocapnia can result in hyperemia and elevated cerebral blood flow. This could be harmful in patients with intracranial pathology such as TBI, where raised intracranial pressure is of concern.20
The Cardiovascular System
Although hypercapnic acidosis inhibits cardiac contractility and reduces systemic vascular resistance, the net impact of hypercapnia is an increase in cardiac output through sympathoadrenal mechanisms. This is associated with increased preload, decreased afterload, improved contractility, and elevated heart rate.21 Oxygen delivery is further augmented in hypercapnic acidosis as a result of a rightward shift in the oxyhemoglobin dissociation curve.
In contrast, hypocapnia has the opposite effect on the oxyhemoglobin dissociation curve and reduces myocardial blood flow through coronary artery constriction. With the increased heart rate and contractility that accompany hypocapnia, this can potentiate cardiac ischemia in acute illness.22
The Immune System and Inflammation
Hypercapnic acidosis suppresses both innate and adaptive immune responses. Specifically, hypercapnic acidosis reduces neutrophil and macrophage migration to septic foci while inhibiting phagocytosis. It further impairs the release of pro-inflammatory cytokines such as tumor necrosis factor alpha, interleukin-8, and interleukin-6.23 Acidosis also impairs lymphocyte and natural killer cell cytotoxicity (Fig. 1). Most importantly, clinically relevant levels of acidosis have been associated with bacterial proliferation in some models of infection. Specifically, acidification of culture mediums to pH 7.20 enhances the growth of Escherichia coli, and this effect has been demonstrated across several species of bacteria.25,26 Whether treatment with appropriate antibiotics mitigates this effect in sepsis remains controversial.25,27
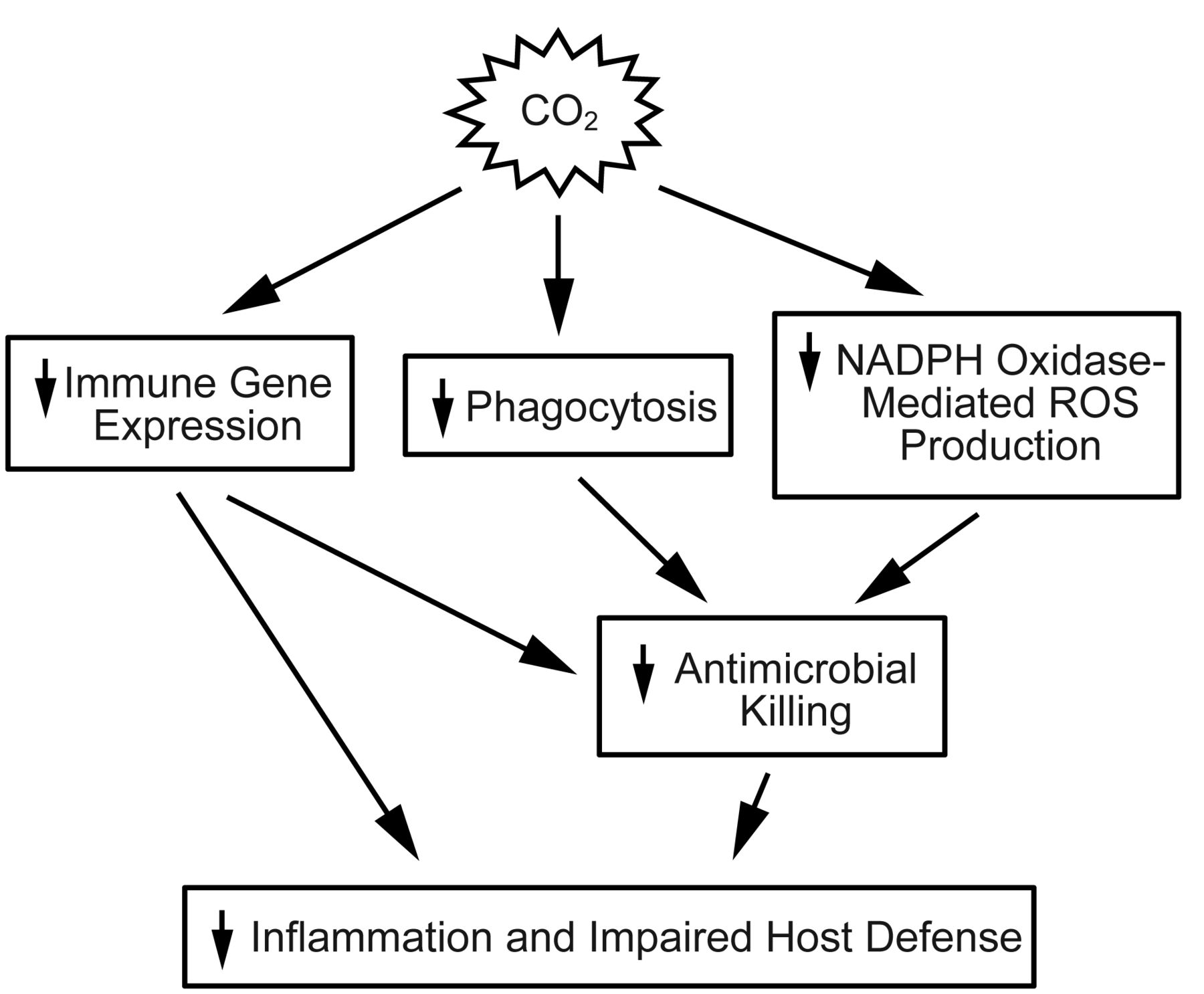
This schematic demonstrates the different mechanisms by which hypercapnia suppresses host defenses. NAPDH = reduced nicotinamide adenine dinucleotide phosphate hydrogen. ROS = reactive oxygen species. From Reference 24, with permission.
Hypocapnia in TBI
In patients with TBI, elevated intracranial pressure is often a concern due to hemorrhage (eg, epidural, subarachnoid, intraparenchymal) and brain tissue edema. The Monro-Kellie hypothesis has been used to illustrate the factors that influence intracranial pressure within the fixed volume of the skull.28 These factors include blood, cerebrospinal fluid, and brain tissue. When a mass lesion (eg, hemorrhage) elevates intracranial pressure, blood and cerebrospinal fluid are displaced to prevent intracranial pressure from rising further. Because cerebral perfusion pressure is equal to the difference between mean arterial pressure and intracranial pressure, cerebral tissue oxygenation becomes impaired as intracranial pressure continues to increase. Moreover, the brain may herniate into compartments of lower pressure to maintain intracranial pressure.29 Once these compensatory mechanisms have been exhausted, intracranial pressure rises at an exponential rate in response to small changes in intracranial volume. If allowed to continue, brainstem herniation occurs, leading to coma and death.
In patients with TBI, cerebral blood flow and oxygen delivery may be substantially reduced to the point where as many as 31% of patients are below the threshold at which their cells experience ischemia and are at risk of cell death.30 Of particular concern is the fact that blood vessels in regions of the brain that have sustained injury demonstrate hyper-reactivity in relation to changes in PaCO2 tension.31 Inducing hypocapnia can thus inflict further damage to these already damaged parts of the brain.
Clinicians will often attempt to lower intracranial pressure medically through promoting venous drainage from the intracranial compartment (eg, loosening cervical spine collars and elevating the head of the patient's bed), administration of hypertonic solutions (eg, 3% saline or mannitol),32 and hyperventilation.33 Inducing hypocapnia through hyperventilation in patients with TBI often occurs to buy time for more definitive interventions. In more severe TBI, this may include surgical management with the insertion of an extraventricular drain that allows for removal of cerebrospinal fluid or decompressive craniectomy.34 Unfortunately, even with optimal medical and surgical management, intracranial pressure will sometimes remain elevated. This can be particularly challenging for clinicians to manage.
In TBI, it has been demonstrated that intracranial pressure varies widely, with hypocapnia exerting only transient reductions but worsening long-term mortality and disability.35,36 Patients who are hyperventilated are more likely to be in a persistent vegetative state or require permanent assistance with activities of daily living if they survive their hospitalization. In contrast, investigators have shown that normocapnia in TBI is associated with less neurologic impairment and better functional independence.36
Clinicians have also used hypocapnia in an attempt to reduce luxury perfusion in brain injury to prevent worsening of cerebral edema. In children with brain injuries, the arteriovenous oxygen content difference (C(A-V)O2) is reduced. Because the C(A-V)O2 is inversely related to cerebral blood flow (CBF; assuming a constant cerebral metabolic rate of oxygen [CMRO2]), a lower C(A-V)O2 suggests excess cerebral blood flow or luxury perfusion (Equation 2).37
 (2)
(2)
Through driving PaCO2 levels down, it has further been proposed that blood would be shunted from uninjured brain tissue to vulnerable regions of the brain where autoregulation is impaired. This has been termed inverse steal.38 These concepts have been discredited, as cerebral blood flow is generally reduced following TBI. Because the blood vessels supplying injured brain tissue also exhibit greater reactivity in response to alterations in CO2, these regions are more vulnerable to ischemic damage. Thus, hypoventilation will shunt blood away from these areas of the brain and preferentially supply normal bran parenchyma.
More sophisticated models of brain injury suggest that regional variability exists in the penumbra surrounding injured brain tissue and that CO2 levels should be titrated to optimize blood flow to these parts of the brain. Data also exist advocating the titration of CO2 to the cerebral metabolic oxygen rate and jugular venous oxygen levels.38 These approaches remain controversial, as they have no proven efficacy in clinical practice. Perhaps of greater importance, titrating CO2 levels to cerebral oxygen demand may prove to be unreliable, as CO2 levels have a direct impact on oxygen consumption through direct stimulation of neuronal excitability.
Investigators have failed to identify a safe threshold of hypocapnia for clinicians to target in treating TBI, but guidelines typically recommend against prophylactic hyperventilation to a PaCO2 of < 25 mm Hg. Clinical practice guidelines state that hyperventilation should only be used as a temporizing measure in cases of elevated intracranial pressure and should be avoided within the first 24 h of injury because cerebral blood flow is often reduced during this time period. Finally, jugular venous oxygenation or brain tissue oxygen tension should be monitored in cases in which hyperventilation is used to minimize the risk of brain tissue ischemia.33
Hypercapnia in ARDS
In patients with ARDS, lung-protective ventilation with pressure-limited and volume-limited ventilation (ie, using a plateau pressure of < 30 cm H2O and VT of 6 mL/kg of predicted body weight) has demonstrated a significant survival benefit.39 Due to the lower VT and consequent reduction in alveolar ventilation, this strategy is commonly associated with the development of hypercapnia. Recognizing that low-VT ventilation confers survival benefits through reducing lung stretch and cyclical collapse of alveoli, clinicians have accepted higher PaCO2 levels in patients receiving lung-protective ventilation,40 thus emerged the concept of permissive hypercapnia.6 In patients receiving lung-protective ventilation with low VT, the resultant hypercapnia may occur within 2 contexts: (1) hypercapnic acidosis, where elevated CO2 levels are accompanied by lower pH; and (2) hypercapnia with normal pH (eg, states of volume contraction, metabolic compensation for respiratory acidosis, etc).
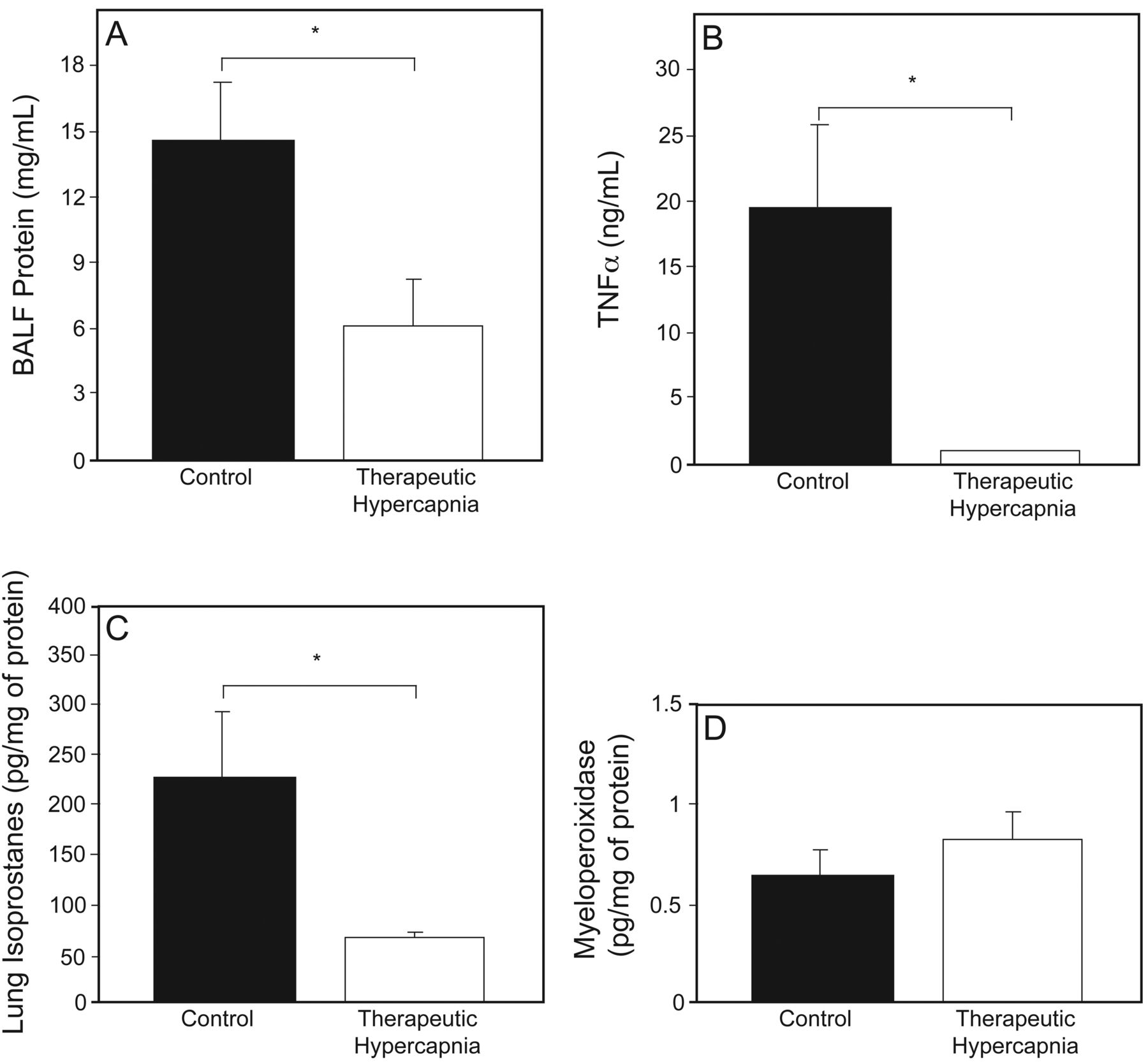
In patients with ARDS, it remains unclear whether hypercapnic acidosis carries survival benefits independent of lung-protective ventilation using low VT. Preclinical investigations have demonstrated hypercapnic acidosis to be associated with reduced levels of protein leak, pulmonary edema, and pulmonary inflammation (Fig. 2). These investigations have further demonstrated hypercapnic acidosis to be protective against free radical-mediated injury while preserving lung compliance.6,41 Conversely, buffering hypercapnic acidosis leads to higher levels of capillary permeability and oxidative damage through increased xanthine oxidase activity42 in lung models of ischemia-reperfusion. In fact, buffering hypercapnic acidosis has been shown to attenuate its protective effects and result in lung injury comparable to controls with normal pH and CO2 levels.43 Similarly, hypocapnia seems to directly potentiate ischemia-induced lung injury, as hypocapnic alkalosis is associated with increased capillary permeability and lung weight, while decreasing lung compliance (Fig. 3).44

In this animal model of lung injury, therapeutic hypercapnia was associated with A: lower levels of protein leak, indicated by decreased concentrations of bronchoalveolar lung fluid (BALF) protein in lung BALF; B: reduced lung inflammation, indicated by lower lung tumor necrosis factor alpha (TNFα); C: less oxidant-induced injury, represented by smaller amounts of lung isoprostane levels; and D: comparable degrees of pulmonary neutrophil content, indicated by equivalent levels of lung myeloperoxidase. Data are shown as mean ± SD. * Statistical significance. From Reference 41, with permission.

After ischemic-reperfusion (IR), capillary permeability and wet:dry ratios were higher in the control and buffered hypercapnia groups in this animal model of lung injury. From Reference 43, with permission.
It is of particular concern, however, that hypercapnic acidosis impairs cell membrane repair and alveolar fluid clearance45 while also suppressing the immune response.24 This is significant, as the majority of ARDS cases result from pulmonary infection.46 Under these circumstances, inhibition of the immune response and impaired cell membrane healing could worsen pulmonary injury and propagate distant organ failure, the main cause of death in patients with ARDS.
Although clinical data reporting the impact of hypercapnic acidosis in ARDS are lacking, some preliminary findings seem to suggest benefits. In 2 randomized controlled trials evaluating lung-protective ventilation, PaCO2 levels remained higher in subjects receiving low-VT ventilation despite having an equivalent minute ventilation to the conventional VT group.39,47 Although PaCO2 levels were higher in subjects receiving lung-protective ventilation in these studies, it is important to note that there was no difference in pH between the 2 groups after 36 h. This is likely attributable to metabolic compensation and the use of buffering agents to treat acidosis.
Interestingly, a secondary analysis of the ARDS Network clinical trial data demonstrated hypercapnic acidosis to be associated with lower mortality in subjects receiving mechanical ventilation with VT of 12 mL/kg of predicted body weight.48 This effect was not observed in subjects who were ventilated at VT of 6 mL/kg of predicted body weight. The investigators theorized that ventilator-induced lung injury occurred to a greater extent in subjects receiving VT of 12 mL/kg of predicted body weight and that these harmful effects were mitigated by hypercapnic acidosis. They further surmised that lung-protective ventilation reduced the impact of ventilator-induced lung injury to the point where the protective effect of hypercapnic acidosis could not be detected. Accordingly, the impact of hypercapnic acidosis in reducing lung injury was diminished by the fact that less lung injury was occurring in these subjects.
Currently, some degree of permissive hypercapnia and associated respiratory acidosis is commonly accepted within the context of mechanical ventilation strategies that target low VT. With the emergence of extracorporeal life support to facilitate lung rest (eg, peak inspiratory pressure of 20–25 mm Hg, PEEP of 10–15 cm H2O, breathing frequency of 10 breaths/min, and FIO2 of 0.3)49 and ultraprotective mechanical ventilation with VT (< 6 mL/kg of predicted body weight),50,51 PaCO2 levels can be titrated to very specific levels. In fact, refractory respiratory acidosis is considered by some institutions to be a criterion for instituting extracorporeal life support.52,53 With the technology available to normalize CO2 levels and allow clinicians to implement ventilation strategies aimed at reducing ventilator-induced lung injury, it remains unclear whether any clinical benefit can be derived from hypercapnic acidosis in preserving pulmonary function and promoting recovery. As these technologies become increasingly utilized in the ICU, it remains to be seen how the role of hypercapnia/hypocapnia will evolve.
Buffering Hypercapnic Acidosis
Profound levels of hypercapnic and metabolic acidosis (pH < 7.10) are associated with adverse physiologic effects.54,55 Particularly, severe acidosis can impair myocardial contractility and reduce cardiac output, leading to hypotension that is refractory to catecholamine infusions. Furthermore, severe acidosis can alter mental status, impair immunologic function, and reduce energy metabolism. Buffering hypercapnic and metabolic acidosis remains common clinical practice, although it is controversial.55
Studies examining the use of sodium bicarbonate infusions in subjects with lactic acidosis have failed to show a benefit in improving pH and suggested many adverse effects to be related to treatment, including hypervolemia, hyperosmolarity, and increased lactate levels.54
Furthermore, sodium bicarbonate infusions have been used to buffer hypercapnic acidosis occurring in the context of lung-protective ventilation. In fact, bicarbonate infusions were permitted in cases in which pH fell below 7.15 in the original ARDS Network clinical trial.39 Once infused, bicarbonate anions combine with hydrogen ions produced by weak organic acids and dissociate to produce CO2. The effectiveness of bicarbonate infusions is dependent on the ability to excrete carbon dioxide. This may be limited in patients with ARDS receiving controlled mechanical ventilation, who have reduced and fixed alveolar ventilation. The CO2 generated from bicarbonate infusion can further worsen cellular acidosis through passively diffusing into cells and reacting with carbonic anhydrase to produce carbonic acid.3,54,55
Alternatively, there may be a role for using tris-hydroxymethyl aminomethane (THAM) to buffer hypercapnic acidosis, as it can easily enter cells to buffer pH and simultaneously reduce carbon dioxide levels.56 Through more effectively correcting pH, THAM can also mitigate the adverse effects of acidosis on the cardiovascular system and restore hemodynamic stability.57
Areas of Uncertainty and Future Research
Several areas of uncertainty surround the role of hypercapnia/hypocapnia in clinical practice. First, hypocapnia is commonly used in TBI to reduce intracranial pressure emergently. Because hypocapnia-induced vasoconstriction carries the risk of inducing cerebral ischemia, further research is required to determine the best way to monitor cerebral tissue oxygen to balance oxygen delivery with the need to reduce cerebral blood volume. Next, although many preclinical studies suggest that hypercapnic acidosis reduces lung inflammation and preserves lung compliance in models of lung injury, future investigations should focus on determining whether hypercapnic acidosis promotes recovery and improves survival independently of low-VT ventilation in patients with ARDS. Furthermore, the utility of buffering hypercapnic acidosis in the context of lung-protective ventilation has been questioned. Finally, it remains unclear whether the possible benefits of hypercapnic acidosis seen in preclinical studies outweigh the potential risks associated with immunosuppression and increased bacterial proliferation.
Summary and Recommendations
Hypercapnia and hypocapnia are common in critically ill patients. Although hypercapnia has been associated with higher cardiac output and improved ventilation/perfusion matching, it has also been shown to suppress the immune response in several models of sepsis and may actually promote bacterial growth.
TBI
Because hypocapnia in the context of TBI confers an increased risk of cerebral ischemia, it is recommended only in emergent situations to reduce intracranial pressure while more definitive (ie, surgical) interventions are being arranged. Sustained hypocapnia is not recommended, as it provides only a temporary reduction in intracranial pressure and is associated with higher rates of mortality and disability.
Permissive Hypercapnia in ARDS
Outside of lung-protective ventilation, hypercapnic acidosis does not have a current indication in clinical practice. Although preclinical studies and retrospective clinical investigations have suggested that hypercapnic acidosis reduces lung injury, its current utility in clinical practice remains unclear, with insufficient data to guide clinicians at this time. Buffering hypercapnic acidosis is recommended only for a specific indication (ie, hemodynamic compromise). In situations in which buffering substantial acidosis (pH < 7.10) is being considered, THAM is recommended over sodium bicarbonate, as it is more effective in mitigating the hemodynamic effects of acidosis and does not increase CO2 levels.
Footnotes
- Correspondence: Eddy Fan MD PhD, Toronto General Hospital, 585 University Ave, PMB 11-123, Toronto, Ontario M5G 2N2, Canada. E-mail: eddy.fan{at}uhn.ca.
Dr Fan presented a version of this paper at the 29th New Horizons in Respiratory Care Symposium: Back to the Basics: Respiratory Physiology in Critically Ill Patients at the 59th AARC Congress, held November 16–19, 2013, in Anaheim, California.
The authors have disclosed no conflicts of interest.
- Copyright © 2014 by Daedalus Enterprises





{kind=link}
{kind=link}
{kind=link}