

Abstract
Inhalation therapy has matured to include drugs that: (1) deliver nucleic acids that either lead to the restoration of a gene construct or protein coding sequence in a population of cells or suppress or disrupt production of an abnormal gene product (gene therapy); (2) deliver peptides that target lung diseases such as asthma, sarcoidosis, pulmonary hypertension, and cystic fibrosis; and (3) deliver peptides to treat diseases outside the lung whose target is the systemic circulation (systemic drug delivery). These newer applications for aerosol therapy are the focus of this paper, and I discuss the status of each and the challenges that remain to their successful development. Drugs that are highlighted include: small interfering ribonucleic acid to treat lung cancer and Mycobacterium tuberculosis; vectors carrying the normal alpha-1 antitrypsin gene to treat alpha-1 antitrypsin deficiency; vectors carrying the normal cystic fibrosis transmembrane conductance regulator gene to treat cystic fibrosis; vasoactive intestinal peptide to treat asthma, pulmonary hypertension, and sarcoidosis; glutathione to treat cystic fibrosis; granulocyte-macrophage colony-stimulating factor to treat pulmonary alveolar proteinosis; calcitonin for postmenopausal osteoporosis; and insulin to treat diabetes. The success of these new aerosol applications will depend on many factors, such as: (1) developing gene therapy formulations that are safe for acute and chronic administrations to the lung, (2) improving the delivery of the genetic material beyond the airway mucus barrier and cell membrane and transferring the material to the cell cytoplasm or the cell nucleus, (3) developing aerosol devices that efficiently deliver genetic material and peptides to their lung targets over a short period of time, (4) developing devices that increase aerosol delivery to the lungs of infants, (5) optimizing the bioavailability of systemically delivered peptides, and (6) developing peptide formulations for systemic delivery that do not cause persistent cough or changes in lung function.
- aerosol viral vectors
- aerosol non-viral vectors
- interfering RNA
- lung cancer
- alpha-1 antitrypsin
- cystic fibrosis (CF)
- vasoactive intestinal peptide
- glutathione (GSH)
- granulocyte-macrophage colony-stimulating factor (GM-CSF)
- insulin
Introduction
There are many advantages to administering medications to the lung as an aerosol. This route of administration provides a more rapid onset of action for short-acting bronchodilators compared with oral therapy, high local drug concentration by delivery directly to the airways, needle-free systemic delivery of drugs with poor oral bioavailability, and pain- and needle-free delivery of drugs that require subcutaneous or intravenous injection. Traditional aerosol therapies consist of short- and long-acting β2-adrenergic agonists, anticholinergics, steroidal and non-steroidal anti-inflammatories, antibiotics, and mucolytics. This paper focuses on newer aerosol therapies for oral inhalation, including: (1) aerosolized gene therapies to treat lung cancer, Mycobacterium tuberculosis, alpha-1 antitrypsin deficiency, and cystic fibrosis (CF); (2) aerosolized peptides to treat asthma, sarcoidosis, pulmonary hypertension, CF, and pulmonary alveolar proteinosis; and (3) aerosolized peptides to treat diseases outside the lung whose target is systemic circulation. The status and challenges facing each of these new applications are presented. Results from clinical and preclinical studies are highlighted, and delivery devices are featured if they appear critical to the success of the application.
Aerosolized Gene Therapies to Treat Lung Diseases
Formulation Options
The goal of aerosolized gene therapy is either to correct the lung disorder with delivery of a functional copy of the aberrant gene to the appropriate target within the lung or to suppress or disrupt production of an abnormal gene product. Viral or non-viral vectors can deliver nucleic acids that lead to permanent expression of a gene construct or protein coding sequence in a population of cells within the lung, thereby reversing or preventing the disease process. Interfering ribonucleic acids (RNAs) can suppress production of the abnormal gene product. One challenge to aerosolized gene therapy is choosing the best approach.
Viral Vectors.
There are a number of viral vectors that can deliver functional genetic material by aerosolization. Viral vectors include retroviruses, lentiviruses, adenoviruses, and adeno-associated viruses. Table 1 summarizes the advantages and limitations of viral vectors that have been tested as possible carriers of genetic material in treating various lung diseases. Details about these vectors have been published previously.1,2
Aerosolized Gene Transfer Agents: Viral Vectors
It is clear from Table 1 that one of the main drawbacks to the use of adenoviral or adeno-associated viral vectors is the high probability of inducing an acute immune response upon initial administration or reduced efficacy following repeat dosing, respectively. This is problematic since aerosolized gene therapy will likely require repeat dosing because mucus clearance mechanisms and/or phagocytes may engulf and destroy the drug vector before it can be taken up by all the targeted cells. In the case of adenoviruses, the deoxyribonucleic acid (DNA) does not integrate into the host genome, and so its effects are transient, lasting only 7–42 d.2 In addition, when treating cancer cells with aerosolized gene vectors, it is important to remember that cancer cells mutate, and tumor-specific treatment agents may need to be altered and reintroduced using a similar vector to treat mutations that become resistant to previous therapies.3
There are additional challenges to aerosolized gene therapy with viral vectors. (1) Delivery is limited to liquid formulations using a nebulizer. There is no dry powder or metered-dose inhaler formulation currently available for any vector-drug combination. However, delivery efficiency is too low with traditional nebulizer devices. Therefore, devices that efficiently deliver gene doses over a short period of time are needed to ensure treatment compliance and reduce the costs associated with gene transfer therapy, which can be extremely expensive. (2) Delivery of the aerosolized drug vector to the lungs of patients with chronic airway obstruction can be non-homogeneous, with the drug vector depositing only in regions of the lungs that are ventilated. Figure 1 shows that as airway obstruction increases, indicated by a lower percent-of-predicted FEV1, distribution of the radioactive aerosol becomes more uneven. Similar non-uniformity in the distribution of a drug vector could make treatment less efficacious. (3) Anatomic, physiologic, and behavioral factors limit delivery of an adequate dose of drug to the lungs of infants. Devices that increase aerosol delivery to the lungs of infants are needed. This is especially important when treating lung diseases such as CF. Identification of infants who are afflicted with CF is now possible at birth, and early gene therapy holds the promise of correcting the abnormality before irreversible lung damage can occur. (4) Vectors whose target is the airway epithelial cell must be able to penetrate beyond the airway mucus barrier. However, the mucus barrier is thick and viscous in adult patients with CF and in patients with COPD. (5) Vectors are needed that recognize receptors on the apical surface of airway epithelial cells. Many vectors recognize only receptors on the basal or lateral surfaces, which are very difficult to access. (6) Gene vectors that transport DNA must deliver DNA to the cell nucleus. To do this, the vector must penetrate the cytoplasmic membrane, overcome cytoplasmic proteases, and penetrate the nuclear membrane.
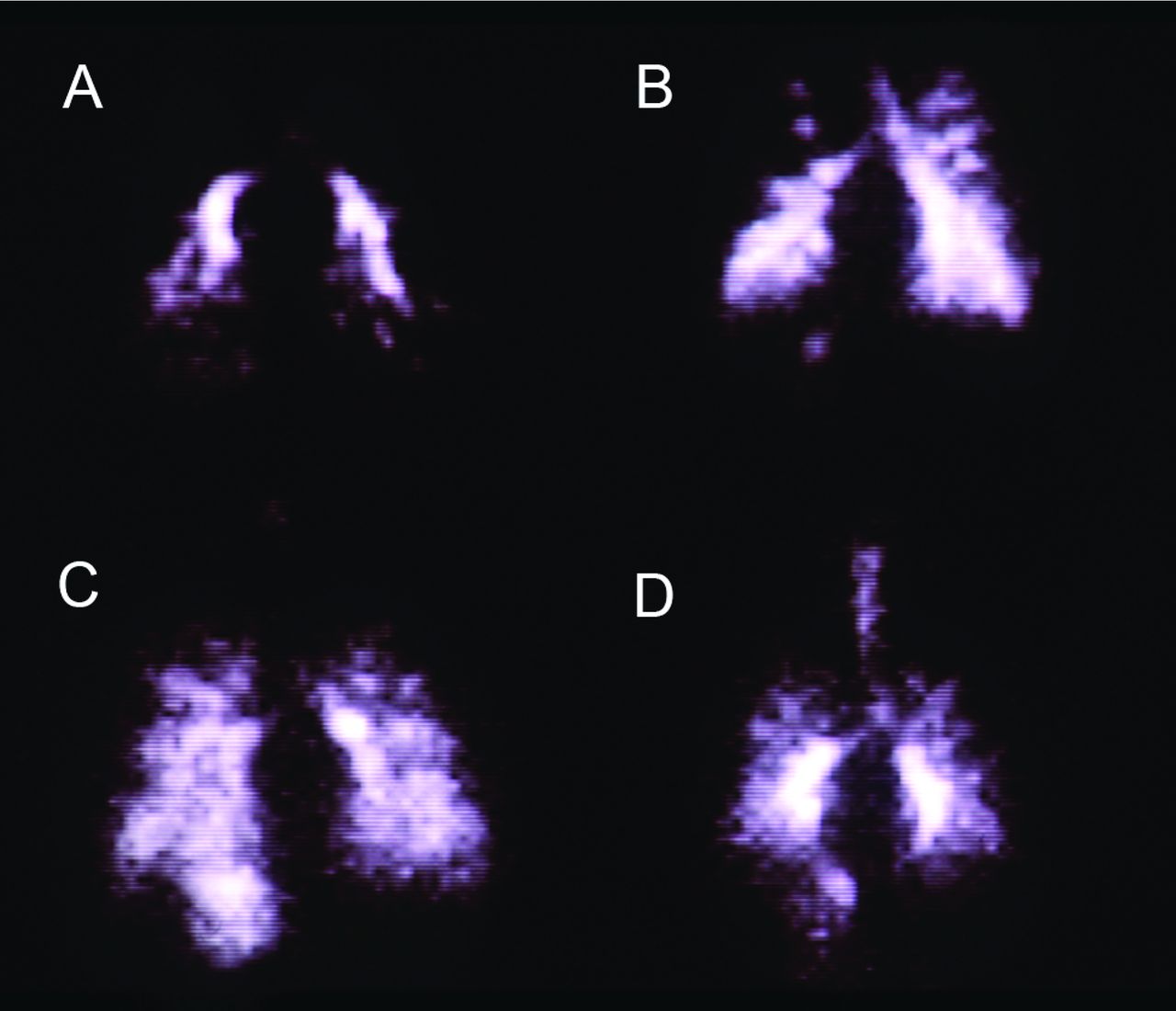
Gamma camera scans showing posterior lung images of 4 adult patients with cystic fibrosis following inhalation of an aerosol containing the radioisotope technetium-99m. Distribution of the aerosolized radioisotope varies significantly with the degree of airway obstruction, as measured by FEV1. A: FEV1 = 25% of predicted. B: FEV1 = 45% of predicted. C: FEV1 = 55% of predicted. D: FEV1 = 60% of predicted. From Reference 4.
Non-viral Vectors.
Non-viral vectors include naked plasmid DNA, cationic lipids, or cationic polymers that bind to negatively charged plasmid DNA and either encapsulate or condense the DNA to generate lipoplexes, polyplexes, and nanoparticle-ligand complexes. Table 2 summarizes the advantages and limitations of non-viral vectors that have been tested as possible carriers of genetic material in treating various lung diseases. Details about these vectors have been published previously.1,2 The simpler composition of non-viral vectors may have an advantage over viral vectors, making re-administration potentially more successful.5,6 They are also cheaper and easier to produce in large amounts compared with viral vectors. Some progress has been made in improving non-viral gene transfer.7 However, there are safety concerns associated with non-viral vectors, including the induction of cytokine responses and innate immune responses by naked plasmid DNA and plasmid DNA-cationic liposome complexes, respectively.8 In addition, transfection (ie, introducing nucleic acids into cells) with polyplexes is low compared with viral vectors. More work is needed to overcome these limitations.
Aerosolized Gene Transfer Agents: Non-Viral Vectors
Interfering RNAs.
Another approach to gene therapy is to inhibit the expression of a specific gene using interfering RNAs. Interfering RNAs include small interfering RNA (siRNA), short hairpin RNA (shRNA), and microRNA (miRNA). siRNA is double-stranded and 20–25 base pairs long (Fig. 2). It can be chemically synthesized and delivered to cells directly or by lipid-mediated transfection. Within the cell cytoplasm, siRNA molecules are picked up by Argonaute proteins (Fig. 2). The Argonaute protein strips away one strand from the siRNA and cleaves any messenger RNA (mRNA) that matches it, destroying it. In this way, the siRNA inhibits the expression of specific genes with complementary nucleotide sequences within the cell.9 The degree of inhibition by siRNAs in mammalian cells is rarely 100%. Therefore, the inhibition of gene expression is usually referred to as a disruption in expression. It is unclear what parameters determine the effectiveness of a given siRNA.10,11
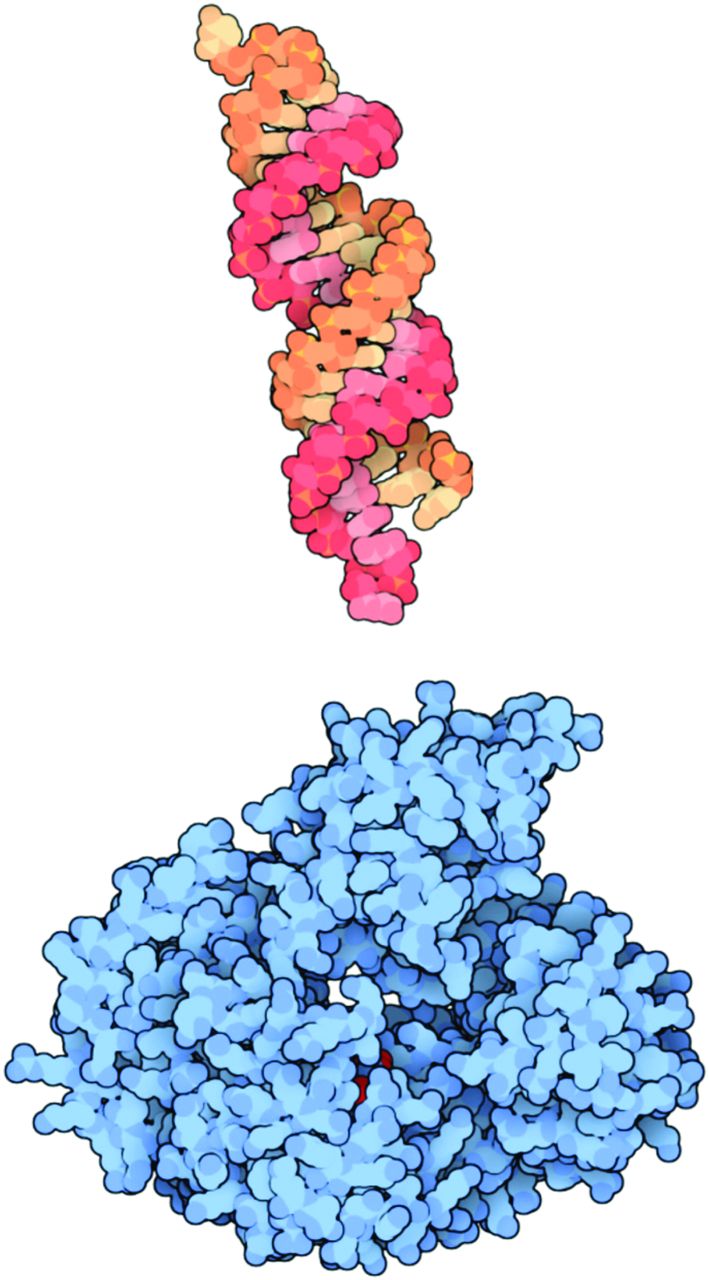
An example of a small interfering ribonucleic acid (siRNA) molecule is shown at the top in orange and red. Once inside the cell, the siRNA molecule is picked up by an Argonaute protein, shown at the bottom in blue. The Argonaute protein strips away one strand from the siRNA and then looks for any messenger RNA within the cell that matches it. If it finds one, it cleaves the RNA, destroying it. From Reference 9, with permission.
Chemically synthesized siRNAs can be purchased from a number of commercial sources, but they are prohibitively expensive.12 shRNAs are a less expensive method for generating interfering RNAs. shRNAs are a single transcript, and their structure can be based on both siRNAs and miRNAs.13,14 miRNAs are single-stranded and appear to inhibit gene expression by pairing directly with an mRNA sequence within the cell and repressing protein translation, which is in contrast to RNA cleavage mediated by siRNAs.12
Unlike siRNAs, shRNAs are synthesized in the cell nucleus and require an expression vector. Once in the cell nucleus, they are further processed and transported to the cytoplasm, where they are assimilated into the endogenous miRNA pathway.15 Thus, shRNAs can be continuously synthesized by the host cell, making their effect much more durable (ie, knockdown of gene expression is longer term) compared with exogenously introduced siRNAs.15 Data from the few studies that have compared the efficacy of RNA interference to that of single-stranded antisense-mediated inhibition of gene expression in mammalian cells indicate that siRNAs and miRNAs are at least as effective as standard antisense oligonucleotides.16–18
Interfering RNA molecules have shown promise in suppressing gene expression, which could be useful in treating cancer. However, this therapy is limited with respect to the extent that molecules can access primary and metastatic target cells.19 In addition, concern has been raised regarding the safety of RNA interference, especially the potential for off-target effects, in which a gene with a coincidentally similar sequence to the targeted gene is also repressed.19
Aerosolized gene therapies with non-viral vectors and interfering RNAs have delivery challenges that are similar to those with viral vectors. Delivery efficiency is likely too low for therapeutic efficacy with traditional nebulizer devices. New devices that deliver adequate doses of the genetic material to lung targets in a short period of time are needed. In addition, non-viral vectors and interfering RNAs face non-uniform distribution within the lungs of patients with airway obstruction, and this may affect treatment efficacy.
For the most part, treatment of lung diseases with inhalation gene therapy is in preclinical development (using cell cultures and animal models) or in phase-1/2 human trials. Aerosolized gene therapies to treat lung cancer, M. tuberculosis, alpha-1 antitrypsin deficiency, and CF are under development. Both the current status of traditional aerosol therapies for these lung diseases and the potential for future gene therapies are discussed below.
Therapies for Lung Cancer
Lung cancer accounts for 23% of total cancer deaths, and non-small cell lung cancer is the most common type of lung cancer worldwide. Although systemic chemotherapies are now customized according to individual biological tumor characteristics, overall survival rates have not improved.20,21 This may be due to low drug concentrations at the tumor site with injection therapy.22,22 It is therefore not surprising that aerosol administration is being tested as a method to deliver higher drug concentrations of systemic chemotherapies to lung tumors while preventing toxic drug concentrations in the systemic circulation. Although a number of drugs have been effective in vitro and in animal models,24–49 inhalation chemotherapies to treat lung cancer have not progressed beyond phase-1/2 clinical trials in humans due in part to toxicities, poor delivery, and lack of efficacy.50–55
Another development designed to improve delivery of inhaled chemotherapy drugs to the lungs is to formulate them as nanoparticles.39,40,47–48 Research is also focusing on liposomal carriers of potent chemotherapy agents such as 9-nitro-camptothecin.46,54 Future clinical trials that integrate safety and efficacy are needed to determine whether chemotherapy via inhalation is a viable treatment approach for lung cancer.
Treating lung cancer through RNA interference could be another approach. The use of siRNAs and shRNAs to treat cancer is still in early preclinical testing. An siRNA has shown promise as a therapeutic agent in inducing apoptosis in myelogenous leukemia cells56 and reduced proliferation in hematopoietic cell lines.57 An shRNA against an oncogenic K-RAS allele delivered by retrovirus demonstrated a reversal of the tumorigenicity of a human pancreatic carcinoma cell line expressing K-RAS in vitro and in vivo.58 Other targets for RNA interference include vascular endothelial growth factor, a critical protein involved in angiogenesis and tumor development59; erbB1, a tyrosine kinase growth factor receptor that is overexpressed in a wide range of cancers60; and the MDR1 gene, which is associated with multiple drug resistance.61 More testing is needed to determine whether these new molecules can be delivered in vivo to cancer cells and if delivery leads to a reduction in gene expression.
Therapies for M. tuberculosis
Tuberculosis is a global problem caused by infection with M. tuberculosis bacilli. At the present time, one third of the world population has been exposed to this bacillus, and 2 million people die every year of tuberculosis.62 Treatment usually involves 2–4 drugs taken for many months and sometimes for years. Failure of a patient to take the medications as prescribed is the most important cause of failure to cure the tuberculosis infection.
Although many investigators have studied numerous antitubercular inhalable formulations, none is yet available in clinics. The latest candidate for inhalation therapy for tuberculosis is siRNA. It has been demonstrated that siRNA uptake into the cytoplasm of lung cells is very efficient and can occur in the absence of vectors or transfection agents.63–66 Taking advantage of this, Rosas-Taraco et al67 investigated whether intrapulmonary delivery of aerosolized XCL1-targeting siRNA could induce suppression of lymphotactin in mice infected with M. tuberculosis. Lymphotactin is one of the chemokines known to participate in the formation of tuberculoid granuloma and is produced by the XCL1 gene. siRNA was delivered to the lungs using an intratracheal MicroSprayer aerosolizer (Penn-Century, Wyndmoor, Pennsylvania). The investigators reported that local pulmonary siRNA therapy resulted in 40–50% protein suppression at 3 and 5 d after treatment (Fig. 3). These findings indicate that it is possible to modulate local lung immunopathology in a murine tuberculosis model using siRNA. A variety of acute and chronic lung diseases are known to have altered expression of cytokines/chemokines or other factors, many of which could be directly involved in lung pathology. Additional studies are needed to determine whether this same gene therapy approach could be used in humans to alter immunopathologies associated with other respiratory diseases.
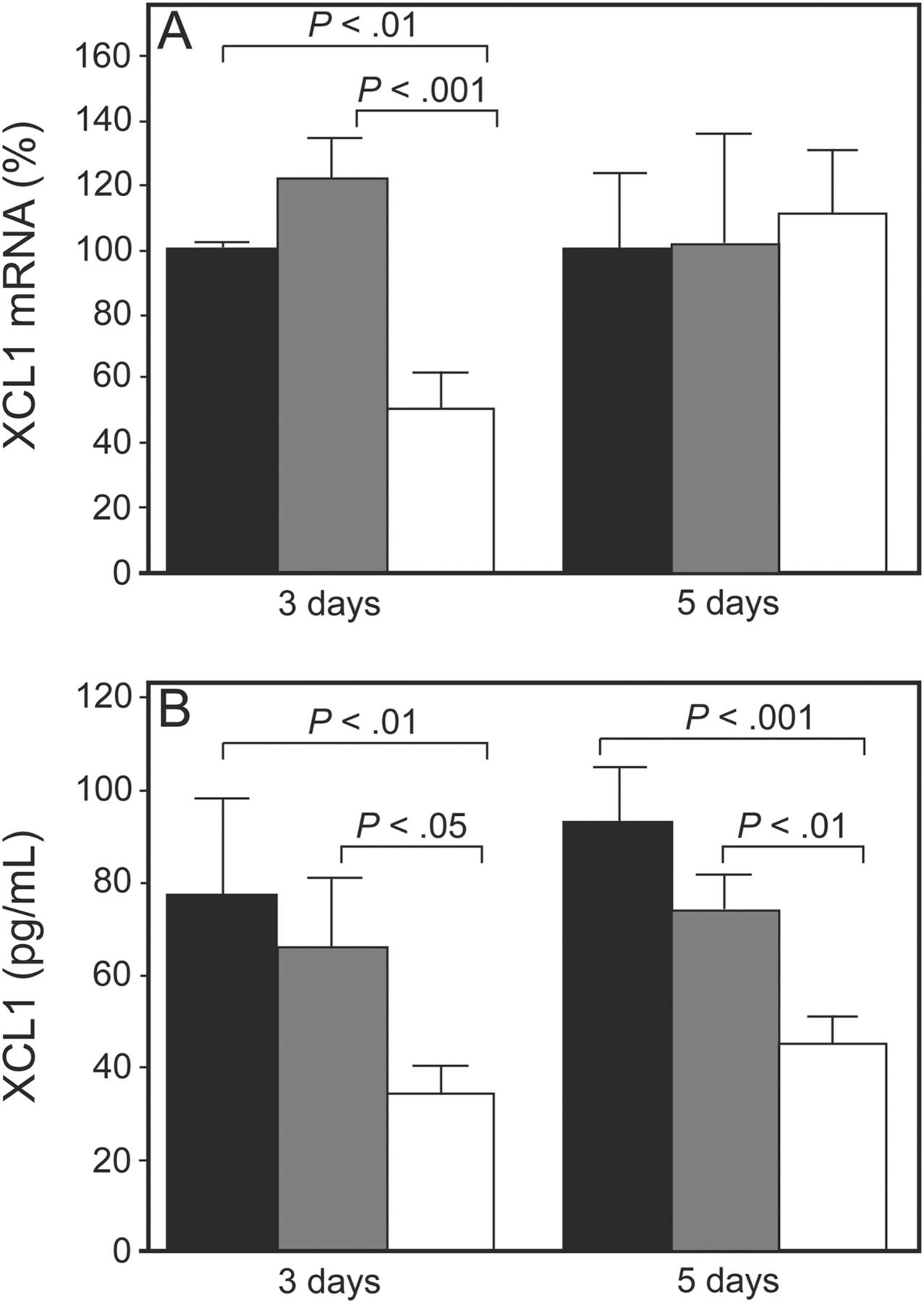
Small interfering ribonucleic acid (siRNA) targeting XCL1 induced transient gene silencing and suppression of protein expression in mice infected with M. tuberculosis. A: XCL1 transcripts (messenger RNA [mRNA]) were reduced by 50% 3 d after a single dose (15 μg/mouse) of XCL1-targeting siRNA (white bars) compared with non-targeting siRNA (gray bars) and phosphate-buffered saline (black bars). Five days after treatment, expression levels of XCL1 transcripts were similar to those in control mice. B: Mice treated with XCL1-targeting siRNA (white bars) showed a 40–50% reduction in XCL1 protein 3 and 5 d after treatment compared with the control groups (black and gray bars). Data represent the mean ± SEM of XCL1 mRNA (%) or XCL1 production from phosphate-buffered saline. From Reference 67, with permission.
Therapies for Alpha-1 Antitrypsin Deficiency
Alpha-1 antitrypsin deficiency, also known as alpha-1 proteinase inhibitor deficiency or alpha-1, is a genetic condition that increases the risk of developing pulmonary emphysema and cirrhosis of the liver. It is caused by mutations in the gene coding for the 52-kDa glycoprotein alpha-1 antitrypsin,68,69 the body's major serine proteinase inhibitor (serpin).70 Alpha-1 antitrypsin deficiency can lead to production of a protein that is dysfunctional as an elastase inhibitor, subsequent unchecked proteolytic activity by neutrophil elastase on lung connective tissue, alveolar destruction, and emphysema. There are ∼100,000 severely deficient individuals in the United States and ∼25 million carriers of at least one gene deficient for alpha-1 antitrypsin.71,72 However, these figures are considered to be significantly underestimated. The most common therapy for alpha-1 antitrypsin deficiency is intravenous augmentation therapy, whereby alpha-1 antitrypsin from the plasma of healthy individuals is purified, and a dose of 60 mg/kg of body weight is intravenously injected on a weekly basis into deficient individuals. This dose appears to produce circulating alpha-1 antitrypsin levels of > 15 μM (80 mg/dL), which seems to protect against lung destruction.73 Although effective, the high cost (∼$80,000/y) and inconvenience of injection therapy for alpha-1 antitrypsin deficiency have led to a focus on direct delivery of alpha-1 antitrypsin to the lung by inhalation. Results from a few phase-1 studies have shown that inhalation of human pooled or recombinant alpha-1 antitrypsin aerosols is safe for human use and raises levels of alpha-1 antitrypsin in plasma and epithelial lining fluid above the protective threshold.74–76 A phase-2/3 multi-center double-blind randomized trial to evaluate the safety and efficacy of inhaled human alpha-1 antitrypsin in 200 subjects with alpha-1 antitrypsin deficiency and emphysema using the eFlow delivery device (PARI, Starnberg, Germany) was recently completed.77 Interim results suggest that the drug is safe and well-tolerated.
It is thought that much of the damage leading to emphysema associated with alpha-1 antitrypsin deficiency occurs in the lung interstitium.78 Thus, it may be necessary to deliver large doses of alpha-1 antitrypsin to the alveolar region of the lung to achieve a therapeutic effect. This will likely not be accomplished by conventional inefficient nebulizers. This has led several investigators to test delivering alpha-1 antitrypsin with inhalation devices that are designed to deposit a high percentage of aerosol in the lungs. Brand et al79 delivered alpha-1 antitrypsin using the AKITA2 APIXNEB inhalation system (Activaero, Gemünden, Germany). This delivery system consists of the eFlow vibrating mesh nebulizer and an electronic unit that controls inhalation flow and inhaled volume by a computerized compressor. Inhalation flow is controlled at a low rate (0.25 L/s) to allow larger particles to bypass areas of obstruction and penetrate more deeply in the lung. Inhaled volume is normalized to the patient's inspiration capacity.
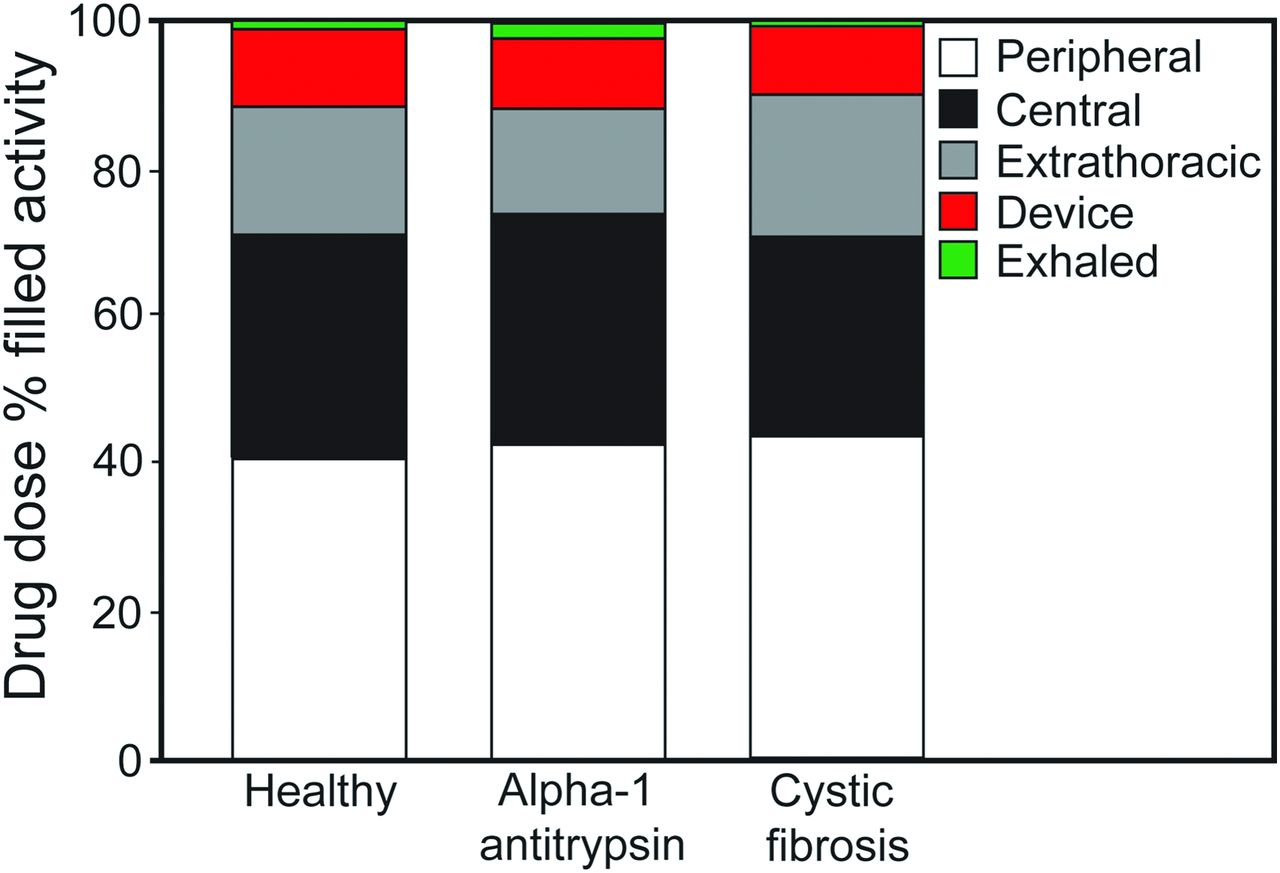
In the study by Brand et al,79 a liquid formulation of alpha-1 antitrypsin was admixed with the gamma-emitting radioisotope technetium-99m, and lung deposition was quantified by gamma scintigraphy (Fig. 4). Twenty subjects (6 healthy, 7 with alpha-1 antitrypsin deficiency, and 7 with CF) inhaled ∼70 mg of radiolabeled alpha-1 antitrypsin in 2 mL of solution. The mass median aerodynamic diameter of the aerosol was 4.0 μm. The total lung deposition of alpha-1 antitrypsin was ∼70% of the nominal dose in the nebulizer, was similar in all study groups, and was unaffected by disease severity. In addition, ∼40% of the nominal dose was deposited in the peripheral-lung region, assumed to be composed primarily of alveoli and small airways (Fig. 5). This study showed that large amounts of alpha-1 antitrypsin were delivered to the lungs of subjects with alpha-1 antitrypsin deficiency or protease-antiprotease imbalance (ie, subjects with CF) in a short period of time using the AKITA2 APIXNEB. Average inhalation times were 6.9, 13, and 8.3 min for healthy subjects, subjects with alpha-1 antitrypsin deficiency, and subjects with CF, respectively.

Examples of ventilation and drug deposition scans of a subject with alpha-1 antitrypsin deficiency. A: Krypton ventilation image. B: Technetium-labeled drug image. Percent drug deposition in the right lung and distribution of the drug within the lung were quantified by determining the lung border on the ventilation scan, registration of that border on the aerosol scan, and further processing of the right-lung region into peripheral- and central-lung regions. From Reference 79, with permission.
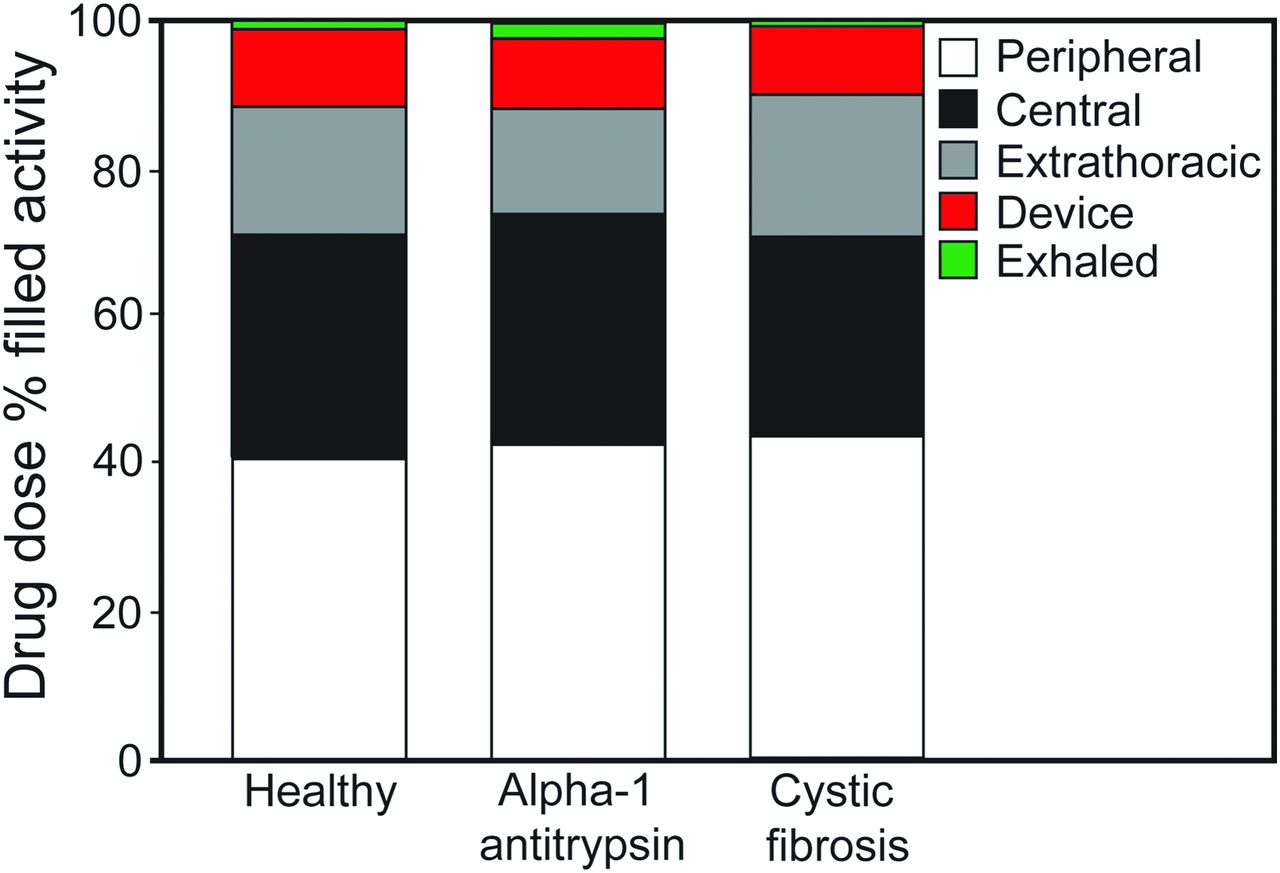
Average drug doses in the peripheral lung, central lung, and extrathoracic airway; residual drug left in the device; and residual drug that was exhaled for the 3 study populations. Individual deposition values were rounded to one decimal place and therefore do not total 100% for each group. From Reference 79, with permission.
Another group of investigators studied the possibility of using the I-neb adaptive aerosol delivery (AAD) system (Philips Respironics, Murrysville, Pennsylvania) to deliver alpha-1 antitrypsin (Fig. 6).80 This delivery system also generates aerosol using vibrating mesh technology. Metering chambers control the drug dosage, delivering a preset volume ranging from 0.25 to 1.4 mL, with a residual of ∼0.1 mL. Medication is delivered into 50–80% of each inspiration based on a rolling average of the last 3 breaths.

I-neb adaptive aerosol delivery system. This delivery system generates aerosol using vibrating mesh technology. Metering chambers control the drug dose, delivering a preset volume ranging from 0.25 to 1.4 mL, with a residual of ∼0.1 mL. Medication is delivered into 50–80% of each inspiration based on a rolling average of the last 3 breaths. Courtesy Philips Respironics.
In an in vitro study, Geller and Kesser80 determined the aerosol characteristics, device efficiency, and delivery time of alpha-1 antitrypsin using the I-neb AAD system during a conventional tidal breathing mode and during target inhalation modes for 6 or 9 s. The nominal dose of alpha-1 antitrypsin in the nebulizer was 25 mg in 0.5 mL. The volume median diameter of the aerosol was 4.4–4.8 μm. The predicted lung dose of alpha-1 antitrypsin as a percentage of the nominal dose was: tidal breathing mode, 56.6% in 7.5 min; target inhalation mode for 6 s, 59.9% in 4.4 min; and target inhalation mode for 9 s, 64.5% in 2.5 min. These results suggest that large amounts of alpha-1 antitrypsin can be delivered to the lungs using the I-neb AAD system.
The development of gene therapy for alpha-1 antitrypsin deficiency is in early preclinical testing. The classic approach of inserting normal genes into cells that are alpha-1 antitrypsin-deficient using viral or non-viral vectors is being investigated. Gene transfer studies utilizing both viral and non-viral vectors have successfully delivered the alpha-1 antitrypsin gene to various target organs and produced alpha-1 antitrypsin in animal models.81–86 In animal studies that specifically targeted the respiratory system, the alpha-1 antitrypsin gene was successfully transferred to airway epithelial cells as demonstrated by mRNA expression, but alpha-1 antitrypsin protein levels in the lung tissue were very low or not determined.87,88 Persistent transgene expression in the airway was demonstrated by transferring the alpha-1 antitrypsin gene to macrophages in vitro using a recombinant adeno-associated viral vector system and then delivering the transduced cells to murine lungs by intratracheal installation.89 The only trial in human airways involved instilling the normal alpha-1 antitrypsin gene in a plasmid DNA-cationic liposome complex into the nasal mucosa of alpha-1 antitrypsin-deficient subjects.90 Increases in alpha-1 antitrypsin levels were observed in nasal lavage samples within days of administration, but returned to baseline by day 14. It is not known if similar increases in alpha-1 antitrypsin levels could be achieved by delivering the normal alpha-1 antitrypsin gene using a similar non-viral vector to the lungs of alpha-1 antitrypsin-deficient patients.
Therapies for Cystic Fibrosis
Approximately 70,000 people worldwide have CF, an inherited autosomal recessive disease. Mutations in the CF transmembrane conductance regulator (CFTR) gene lead to loss of chloride, abnormal sodium and water transport, impaired mucus removal, obstructed airways, chronic infection, and end-stage lung disease. The goal of aerosolized gene therapy is to restore the function of the CFTR and normal chloride channel function in the lungs. Aerosolized gene therapy for CF has had some success in clinical trials. Twenty-four clinical trials with aerosolized gene vectors have been carried out since the cloning of the CF gene in 1989. There have been 9 trials with adenoviruses. Low-level gene transfer was achieved in some subjects, but administration resulted in lung inflammation and induced humoral and cellular immune responses, affecting the efficacy of re-administration.91 Six clinical trials have been carried out with the adeno-associated virus 2 serotype. Acute administration appeared to be safe,92 but there was no improvement in lung function, and repeat dosing does not appear to be possible due to the development of an anti-viral immune response.93 Nine clinical trials evaluated non-viral gene transfer in subjects with CF,91 but none of the trials was designed to assess clinical efficacy. A previous review article provides detailed information about these clinical trials, what vectors were used, and their drawbacks.6
The UK Cystic Fibrosis Gene Therapy Consortium (http://cfgenetherapy.org.uk) is currently conducting the only active CF gene therapy clinical trial. This is a multi-dose double-blinded placebo-controlled clinical trial using the non-viral cationic lipid formulation GL67A (Genzyme, Cambridge, Massachusetts) with specific modifications.6,94 Subjects will be treated with 12 monthly doses delivered by the AeroEclipse II breath-actuated nebulizer (Trudell Medical, London, Ontario, Canada).95
Aerosolized Peptides to Treat Lung Diseases
Several peptides have been proposed as possible pharmacologic agents for treating respiratory diseases, and because of the many advantages to aerosol therapy versus systemic or oral therapy, the preferred route of administration is by inhalation. Peptides that are being tested include vasoactive intestinal peptide to treat asthma, pulmonary hypertension, and sarcoidosis; glutathione to treat CF; and granulocyte-macrophage colony-stimulating factor (GM-CSF) to treat pulmonary alveolar proteinosis. Findings from a number of studies and clinical trials that have been performed with inhaled vasoactive intestinal peptide agonists, glutathione, and GM-CSF are summarized below.
Vasoactive Intestinal Peptide
Vasoactive intestinal peptide contains 28 amino acid residues, is an inhibitory neurotransmitter of the nonadrenergic and noncholinergic autonomic nervous system,96 and is also a T helper 2 cytokine.97,98 It is highly expressed in the lungs and nasal mucosa. Its type-1 receptor (VPAC1) is most abundant in lung tissue and T lymphocytes, and its type-2 receptor (VPAC2) is found in smooth muscle, mast cells, and the basal parts of the lung mucosa.99
The precise role of vasoactive intestinal peptide in the pathogenesis of asthma is not clear. However, its deficiency in the airways of subjects with severe asthma has been reported.100 It has been shown that vasoactive intestinal peptide plasma levels are much lower in subjects with asthma during severe exacerbations compared with healthy individuals.101 It also exerts potent bronchodilator effects, which are independent of adrenergic and cholinergic receptors and cyclooxygenase.102 Interestingly, Lindén et al103 demonstrated that inhalation of a selective VPAC2 agonist (Ro 25-1553) produced a rapid-onset but short-duration bronchodilator effect in 24 subjects with moderate stable asthma. Because vasoactive intestinal peptide affects T helper 2 lymphocyte cells, regulatory T cells, mast cells, eosinophils, neutrophils, and smooth muscle cells,104,105 it is now theorized that a longer-acting aerosolized agonist of this peptide might be used for both maintenance and treatment of exacerbations in patients with asthma.
Vasoactive intestinal peptide is also a key mediator in the pathway leading to pulmonary arterial hypertension,106 with frequent gene alterations identified in subjects with pulmonary arterial hypertension.107 It also causes a 50-fold more potent vasodilation compared with prostacyclin in both pulmonary and systemic arteries.108 Leuchte et al109 demonstrated that aviptadil, an inhaled vasoactive intestinal peptide analogue, induced temporary vasodilatation of the pulmonary vessels and a decrease in the right-heart load in 20 subjects with pulmonary hypertension of different origins, including pulmonary arterial hypertension (n = 9), pulmonary hypertension associated with chronic right-lung disease (n = 8), and pulmonary hypertension with prior thromboembolism to the lung (n = 3). Aerosol was generated by OPTINEB-ir (NEBU-TEC, Elsenfeld, Germany), and the effects of inhaled vasoactive intestinal peptide were observed over a 60-min period.
Inhaled vasoactive intestinal peptide agonists may also have an impact on sarcoidosis because results from a clinical trial showed that vasoactive intestinal peptide nebulized with the OPTINEB-ir for 28 d reduced regulatory T cells and down-regulated the inflammatory status of subjects with sarcoidosis.110 No adverse effects were reported after any of these trials. These results support the potential therapeutic role of inhaled vasoactive intestinal peptide agonists in treating asthma, pulmonary hypertension, and sarcoidosis. More human trials are needed to confirm these early results.
Glutathione
Processes associated with CFTR mutations in patients with CF facilitate excess oxidants in the airways. Glutathione is a naturally occurring tripeptide and a major extracellular antioxidant. Normally, glutathione concentration in the epithelial lining fluid of the lungs is high,111–113 but is depleted in CF.112–114 Early studies investigated whether glutathione inhalation could improve glutathione levels in the lungs. One study showed improvement in alveolar glutathione levels and lung function in subjects with CF after inhalation of glutathione, but no antioxidant effects.113 Several follow-up studies also showed improved lung function after glutathione inhalation.115,116 However, a previous multi-center placebo-controlled randomized trial showed only a minimal improvement in FEV1 (pre-post difference between baseline and end-of-trial FEV1 absolute values) after twice-daily inhalation of glutathione for 24 weeks.117 Placebo and glutathione were both administered by the eFlow nebulizer system. The study population consisted of 153 subjects. The absolute change from baseline FEV1 after 3 months was significantly higher in the glutathione-treated group compared with the placebo-treated group, but this difference was not maintained after 6 months of treatment. In addition, differences in percent-of-predicted FEV1 were not significantly different between the 2 groups at any time point (Fig. 7), and treatment did not diminish oxidative stress or improve anti-inflammatory effects. This was similar to results reported in the authors' previous study.113 There was no reduction in the rate or time to pulmonary exacerbation or improvement in quality of life with treatment. The lack of effect did not appear to be related to poor delivery of glutathione to the lungs because measurements of glutathione and metabolites in sputum at steady state demonstrated significant levels. These results challenge the concept that inhalation of large doses of glutathione may be helpful in mitigating oxidative or inflammatory imbalance in CF.

The absolute change in FEV1 from baseline in glutathione (GSH)-treated subjects was, on average, slightly higher than in the placebo group (open circles) and reached statistical significance at 3 months (A), but not when expressed as percent of predicted (C). Statistical differences were not apparent at 6 months. The Kaplan-Meyer plot shows no difference between drug- and placebo-treated groups in terms of time to first pulmonary exacerbation during the treatment period (B). At all times after the start of treatment, the pre-post differences in free and total GSH in the sputum were significantly higher in the drug-treated subjects (D), indicating good delivery of drug to the lungs. From Reference 117, with permission.
Granulocyte-Macrophage Colony-Stimulating Factor
Pulmonary alveolar proteinosis is a rare condition characterized by the accumulation of lipoproteinaceous material within air spaces.118 Greater than 90% of all reported cases of pulmonary alveolar proteinosis have previously been considered idiopathic but are now thought to be due to a lack of GM-CSF activity in the alveolus, resulting in alveolar macrophage dysfunction and surfactant protein accumulation. Reduced GM-CSF activity has been recently attributed to the presence of a neutralizing autoantibody against GM-CSF protein,119–122 and the condition has been reclassified as autoimmune pulmonary alveolar proteinosis. Typically, the standard of care for pulmonary alveolar proteinosis is whole lung lavage, which is an invasive procedure and does not always lead to a complete and durable resolution of the abnormalities. Reed et al123 showed in the late 1990s that aerosolized GM-CSF ameliorated pulmonary alveolar proteinosis in GM-CSF-deficient mice. These findings suggested that aerosolized GM-CSF might be a suitable alternative to whole lung lavage, and results from a phase-1 pilot study, one clinical trial, and one report of a retrospective case series of subjects who were treated with aerosolized GM-CSF support this notion.124–126 In the larger study performed by Tazawa et al125 (35 subjects), subjects inhaled high (125 μg twice daily) and low (125 μg once daily) doses of recombinant human GM-CSF every other week over 2 sequential 12-week periods. In the smaller retrospective study,126 12 subjects inhaled recombinant human GM-CSF (250 μg twice daily) every other week for various time periods between 1999 and 2003. In both studies, aerosol was generated by the PARI LC Plus nebulizer, and the majority of subjects showed significant improvement in their alveolar-arterial oxygen difference with no serious adverse events. In the study by Tazawa et al,125 24 of 35 subjects responded to inhaled GM-CSF with an improvement of at least 10 mm Hg in their alveolar-arterial oxygen difference during the treatment period (Fig. 8). In the study by Wylam et al,126 11 of 12 subjects showed significant improvement in their pulmonary function, and gas exchange improved significantly in 10 subjects. These data suggest that treatment of pulmonary alveolar proteinosis with aerosolized GM-CSF is feasible, safe, and effective.

A: Overall mean ± SE alveolar-arterial oxygen difference (P(A-a)O2) for all subjects receiving inhalation therapy with granulocyte-macrophage colony-stimulating factor (GM-CSF). * P < .05; ** P < .001. NS = not significant. B: Mean ± SE change in P(A-a)O2 in 24 of 35 subjects who responded to inhaled GM-CSF during each trial period. * P < .05; ** P < .01. A response was defined as a subject who had improvements in P(A-a)O2 of at least 10 mm Hg during the treatment period (weeks 12–36). From Reference 125, with permission.
A previous report by Luisetti et al127 showed that the AKITA2 APIXNEB nebulizer system aerosolized 96% of a recombinant GM-CSF filling dose, and based on the International Commission on Radiological Protection (ICRP) deposition models, it was predicted that 80.4% of the filling dose would deposit in the lungs. No studies have been performed that compare the AKITA2 APIXNEB nebulizer system and the PARI LC Plus nebulizer used in the previous studies to treat pulmonary alveolar proteinosis.
Aerosolized Peptides for Systemic Drug Delivery
The rationale for delivering drugs through the lungs into the systemic circulation by aerosol has been reviewed previously.4 Peptides that have been tested for systemic delivery include interferon β to treat multiple sclerosis; leuprolide acetate to treat prostatic cancer, infertility, and postmenopausal breast cancer; calcitonin to treat postmenopausal osteoporosis; growth hormone-releasing factor to treat pituitary dwarfism; and insulin to treat diabetes. Of these drugs, only calcitonin and insulin have progressed to commercialization.
Calcitonin
Salmon calcitonin is available as Miacalcin nasal spray (Novartis Pharmaceuticals, East Hanover, New Jersey). It is used to treat postmenopausal osteoporosis in females > 5 y after menopause.128,129
Insulin
Insulin has been extensively tested for aerosol delivery through the lungs to treat diabetes and has had some commercial success. An estimated 370 million people worldwide have diabetes,130 and the majority of these have non-insulin-dependent diabetes mellitus, or type-2 diabetes. The majority of patients with type-2 diabetes will eventually need to administer insulin subcutaneously as often as 4 times/d (ie, before meals and at bedtime) to achieve good glucose control. Because injection hurts, compliance with treatment is often reduced. More importantly, patients who would benefit from intervention with insulin treatment decline early treatment because of the pain and inconvenience associated with injection.
A number of devices and formulations for pulmonary delivery of insulin have been tested,131 but until very recently, the Exubera pulmonary insulin delivery system (Pfizer Pharmaceuticals, New York, New York, and Nektar Therapeutics, San Carlos, California) was the only device and formulation for inhaled insulin to be cleared by the FDA. Exubera was cleared in 2006 for use in adults with both type-1 and type-2 diabetes and became commercially available soon after.132,133 By early 2007, 2 additional devices and formulations were in phase-3 testing: the AERx insulin diabetes management system (Novo Nordisk, Bagsvaerd, Denmark, and Aradigm, Hayward, California) and the AIR inhaled insulin system (Eli Lilly, Indianapolis, Indiana; and Alkermes, Cambridge, Massachusetts).134–136 However, between October 2007 and May 2008, production of all 3 products was discontinued. The challenges in developing inhaled insulin and the reasons why commercialization with the Exubera product was discontinued have been recently reviewed.137
A second-generation delivery system and formulation for inhaled insulin were developed by MannKind (Valencia, California), and in June 2014, the company received clearance from the FDA to market their product for mealtime glucose control only. MannKind's delivery system is called Afrezza. It is a pocket-size drug-device combination product (Fig. 9). Insulin microparticles (Technospheres) are introduced into the device in an aspirin-like tablet, which is crushed when the device is closed, releasing drug as a dry powder. In a previous study, investigators found that 90% of Technosphere insulin particles (human regular insulin loaded onto a fumaryl diketopiperazine molecule) were in the respirable range, with a mean particle diameter of 2.5 μm.138 Bioavailability of this new formulation is estimated to be 24–28% of the subcutaneously injected dose.139 A previous review based on a MEDLINE search of studies relevant to Technosphere insulin concluded that it has a pharmacokinetic profile suitable to meet prandial insulin needs in patients with diabetes.140

The Afrezza, a second-generation device for delivering insulin. It is a pocket-size drug-device combination product. Insulin microparticles (Technospheres) are introduced into the device in an aspirin-like tablet, which is crushed when the device is closed, releasing drug as a dry powder. Courtesy MannKind.
To date, Afrezza has not shown any safety issues. Like the Exubera product, there have been reports of coughing during and after inhalation, but coughing is not persistent and appears to be predominant in the early days of treatment.139 Potential effects from insulin remaining in the lungs after inhalation appear to be less than with the Exubera product because insulin in the Afrezza product has a shorter residence time.141 The long-term risks have yet to be determined. The MannKind group did not test Afrezza in subjects with COPD or asthma, and the FDA excluded clearance for the drug in those 2 patient populations.
Another second-generation device for aerosol delivery of insulin is under development. This is the Dance-501 insulin inhaler (Adagio) developed by Dance Biopharm (Brisbane, California). The Dance-501 contains a liquid formulation of natural human insulin instead of a dry powder. Insulin aerosol is dispensed through a small, silent, handheld electronic device in 1–3 breaths (Fig. 10). This product has completed a phase-1/2 trial in subjects with type-1 diabetes and a phase-2 trial in subjects with type-2 diabetes. Results from these trials are unpublished. The company plans to pursue regulatory approvals for mealtime glucose control only for type-2 diabetes in adults in the European Union, United States, and China.

The Dance-501 insulin inhaler, a second-generation device for delivering insulin. The device contains a liquid formulation of natural human insulin instead of a dry powder. Insulin aerosol is dispensed through a small, silent, handheld electronic device in 1–3 breaths. Courtesy Dance Biopharm.
Summary
New applications for aerosol delivery are being developed. These include: (1) aerosolized gene therapies to treat lung cancer, M. tuberculosis, alpha-1 antitrypsin deficiency, and CF; (2) aerosolized peptides to treat lung diseases such as asthma, sarcoidosis, pulmonary hypertension, CF, and pulmonary alveolar proteinosis; and (3) aerosolized peptides to target the lung as an alternative route for administration to the systemic circulation. The status and challenges facing each of these new applications have been discussed.
Aerosolized Gene Therapies to Treat Lung Diseases
In summary, successful correction of lung diseases with inhaled gene therapy remains elusive. Many new approaches are being tested, but few have met with success. A number of challenges must be overcome before aerosolized gene therapies become a reality. These include: (1) selecting the best approach for treating lung diseases by gene therapy (ie, choosing to correct the disease by delivery of a copy of a normal functioning gene or by delivery of an interfering RNA that will inhibit expression of the target gene), (2) developing formulations that are safe and effective for acute and chronic administrations in humans, (3) delivering the genetic material beyond the airway mucus barrier and cell membrane and transferring the material within the cell or to the cell nucleus, (4) improving aerosol delivery to infants, (5) delivering the aerosol dose efficiently in a short period of time to improve compliance and reduce cost.
Aerosolized Peptides to Treat Lung Diseases
Early studies and results support the potential therapeutic role of inhaled vasoactive intestinal peptide agonists in treating asthma, pulmonary hypertension, and sarcoidosis. Similarly, results from several studies in humans suggest that aerosolized GM-CSF might be a suitable alternative to whole lung lavage for treatment of pulmonary alveolar proteinosis. However, more human trials that are randomized and placebo-controlled and that include large numbers of subjects are needed to confirm these early results. Further studies with aerosol delivery systems that carry large doses of peptides to the lungs and alveolar region in a short period of time are also required. Results from a number of clinical trials challenge the concept that inhalation of large doses of glutathione may be helpful in mitigating oxidative or inflammatory imbalance in CF.
Aerosolized Peptides for Systemic Drug Delivery
In 2008, it seemed that the notion of systemic drug delivery to treat millions of patients with diabetes by aerosolization was not achievable. However, to date, there are 2 new formulations/devices for inhaled insulin in late-phase clinical trials (Dance-501 insulin inhaler) or awaiting FDA approval (Afrezza). To ensure satisfactory outcomes and patient acceptability of systemic peptide therapy by aerosolization in the future, it is clear that: (1) the bioavailability of expensive peptides like insulin needs to be optimized; (2) formulations are needed that do not produce persistent cough or changes in lung function and that are safe for acute and chronic administrations; (3) aerosol delivery devices should be small, portable, and easy to use and deliver the drug dose efficiently in a short period of time.
It is likely that aerosolized gene therapies will be developed as liquid formulations and delivered by new improved liquid inhalation systems, including the LC Plus and LC Star breath-enhanced nebulizers (PARI), the AeroEclipse II breath-actuated nebulizer (Trudell Medical), and vibrating mesh nebulizers such as the eFlow (PARI), the AKITA2 APIXNEB (Activaero), and the I-neb AAD system (Philips Respironics). The vibrating mesh nebulizers require specific cleaning procedures that must be followed for long-term use. Aerosolized peptides are being developed as both liquid formulations and powders. A liquid nasal spray formulation for calcitonin (Miacalcin) and a powder formulation for inhaled insulin (Afrezza) have been approved by the FDA and are being commercialized. A liquid formulation for inhaled insulin (Dance-501 insulin inhaler) is under development. Although many of these therapies are only in early testing, respiratory therapists and other caregivers should remain informed about their development in the event that they become available for use in the future. Once they are available to patients, caregivers should follow the directions for use and cleaning included in the package inserts. They should also be alert to possible adverse effects especially in terms of changes in patients' pulmonary function parameters.
Discussion
Willson:
Beth, that was terrific. I wondered as I sat here about the potential of sensitization with inhalation of peptides in the lung. We actually use inhaled insulin, so that question came up with those kids as well. Are there any data on that? Do you have any theoretical concerns about that?
Laube:
Do you mean sensitizing as in making them allergic?
Willson:
Yes.
Laube:
I have not come across any data in that regard myself, but there've been issues with lung function effects as I mentioned, particularly with insulin. But I have not heard about anybody becoming allergic. Perhaps Bruce has had some experience?
Rubin:
We actually ran the only approved clinical trial of inhaled GM-CSF in children with alveolar proteinosis due to a GM-CSF receptor defect. We used the AKITA device. It was stopped because there was development of antibodies that prevented not only continued effect but also put the subjects at some risk.
* Fink:
Great presentation, Beth. In terms of siRNAs, I know that people have had good success in being able to get transfer through the cell membrane. What do you perceive as the challenges of getting through the lung fluids to get to the cells to make the transfer, and are there any strategies and developments to handle those barriers?
Laube:
I didn't mention that as an issue, but it's very important because we can have the best interfering RNAs, but if the aerosol cannot get through the mucus barriers and into the cells, then it will have no effect. That's been one of the problems in CF gene transfer; just getting through the mucus barrier has been quite difficult. I surmise that that is something that will have to be addressed with the interfering RNA delivery systems as well. We have to figure out a way to get through the mucus. There are people who are working on new formulations that can slip through the matrix of the mucus barrier, get through the holes, and get past it. So there's a possibility that if we reformulate properly, that might happen, and there are other people who say that maybe if we used some kind of surfactant additive or something like that, the barrier might be more penetrable as well. It's a very difficult problem to solve.
Restrepo:
Do you anticipate that these 2 new products for insulin delivery will be much cheaper than the one that originally failed? That the cost barrier will be overcome?
Laube:
Yes, the companies seem to have learned that lesson, that the economy of the product was very important in the failure of Exubera. They are saying that these products will be close to the cost of injectable insulin.
† MacIntyre:
I remember being involved with the inhaled insulin safety issue, and the diffusing capacity of the lung (DLCO) was of concern to us. Now, it wasn't much of a drop over 1.5 or 2 years, but it appeared to be real. And the question came up of whether this is a phenomenon that plateaus, you lose 10% of your DLCO capacity, and it stays that way the rest of your life, or do you indeed lose 5–10% of it per year, in which case, in 10 years, you might be a respiratory cripple? I know you don't have the answer, but clearly these sorts of products will have to be followed very carefully for a long period of time when they go on the market. This will be a burden, but an important one, on the developers.
Laube:
The MannKind people say it could be cleared any day by the FDA. In the publications that are available of their clinical trials, they had no problems with DLCO.
† MacIntyre:
How long did they go out?
Laube:
I think they went out a year.
† MacIntyre:
It took over a year in the other one.
Laube:
With the Exubera product, they saw it in 6 months.
† MacIntyre:
Yes, but we weren't really sure. We hadn't really convinced ourselves that it was real.
Laube:
And certainly, it was reversible. So they showed that if they stopped taking the drug, everything went back to normal.
† MacIntyre:
Which is important.
Laube:
But they don't seem to have these issues with the MannKind product or the Dance product.
† MacIntyre:
But you're going to need to follow these people for a long time.
Laube:
Absolutely.
* Fink:
With the Exubera product, the DLCO was not very well quality-controlled. Did they really see those changes in DLCO, and was it poor quality control? Talking to pulmonary function experts who worked with a number of different insulins, it seems that DLCO is a fatal flaw because there's a lot of variability in test methods and reliability between sites. If you don't use simulators and you don't have good quality control, you end up getting erroneous results. It's not clear from the Exubera data whether that was the case in the 2% changes they saw. As I recall, the DLCO level reverted over time rather than getting worse.
Berlinski:
I just had a question regarding CF and gene therapy. I think that when this all got started more than 20 years ago, there was a real need for gene therapy in CF. And I have to tell you, I was surprised coming back from another conference a couple weeks ago that people are still working on gene therapy in CF in light of current developments of molecules that correct the defect. Why do you still see a need? What are your thoughts?
Laube:
Well, it's a mutation that's well known, and if you could reverse that and have normal gene expression, I think that would be very desirable. I think you can probably count on two or three fingers the number of studies that are being conducted today looking at gene therapy for CF. The major emphasis these days is on antibiotic therapies and improving the symptoms, inflammation, and infections that come along with the disease. That seems to be what is leading to the increased longevity of these patients, which is significant, but it's all occurring because of antibiotic therapies. So I think right now that's where most people are focusing their efforts in treating CF, and very little is going into gene therapy today.
Berlinski:
I was referring specifically to drugs that correct CFTR function when we already have one drug (namely, ivacaftor) that is cleared by the FDA to correct function in a specific group of patients. There are more drugs that restore function in the pipeline, and they are undergoing phase-2/3 trials. If that becomes a reality, at least for CF, there shouldn't be any need to correct the gene when you've restored the function.
Laube:
I think ivacaftor is a potentially terrific drug for CF, I agree. But again, there are a few centers that are still very much interested in the possibility of reversing the gene completely and returning it to normal.
O'Malley:
Ivacaftor and the other oral drugs in the pipeline you're talking about will potentially help about 90% of the people with CF. The ultimate goal and desire in CF research is to treat 100% of people with CF, and I'm hopeful that gene therapy will be a possibility.
‡ Suggett:
I have a question, or maybe just a comment, about devices. The CF study1 in the United Kingdom was Davies' work in Oxford. I think it's important, as others have mentioned, that we choose the right delivery devices based on the nature of the formulation and the delivery objective. In that study, I think they looked at a number of different devices, including the I-neb and the eFlow with the AKITA. Following review, they decided that a jet nebulizer with breath-actuated technology was actually the best one in that circumstance. So it's not always the case that you need the higher cost technology; it does need to be looked at on a case-by-case basis.
Laube:
That's an excellent point. In the ongoing CF studies in the United Kingdom,2,3 I think they're using the AeroEclipse II.
‡ Suggett:
Yes, they are.
Ruppel:
I have a follow-up question to what Jim (Fink) asked about the interfering RNA. You alluded to how long that effect lasts. Can you give us a time scale as to how long?
Laube:
In the literature4 I read, it's short in the sense that the siRNA is very susceptible to degradation within the cell and can degrade within a few minutes. What's nice about the shRNA is that it seems to have a longer time period. It's not as quickly degraded, so it tends to stay in the pathway doing its job, pairing up with the mRNAs that are the targets and eliminating them for a much longer period of time, but it does seem to be minutes.
Corcoran:
Are there any general rules about aerosolizing or getting these bigger molecules out? Is there one type of nebulizer that works better for proteins, or is dry powder a better option? Any general rules like that? That seems to be more and more what people are asking to actually deliver in inhaled form.
Laube:
For proteins in general?
Corcoran:
Just for bigger molecules—proteins would fall into that category.
Laube:
Well, with most of the traditional jet nebulizers, we have to be a little careful and make sure that they don't make the product nonviable. But when I was working on CFTR gene therapy with our group and trying to deliver this to animals, we checked viability after jet nebulization, and the proteins held up okay. These proteins were not as susceptible to degradation as I thought they would be. Nevertheless, I do think that you have to make sure that these products have not been affected by the delivery system before you do any kind of large trial. I don't think there are any general rules about this—proteins come in all different sizes and could behave somewhat differently in nebulizers and in other formulations. So you need to be sure that you check viability before and after delivery.
Hill:
I just wonder if you could clarify one thing I was confused about earlier in your talk. You were discussing the XCL1 pathway in relation to tuberculosis and a therapeutic approach that would inhibit granuloma formation. It's not clear to me why that would be of value in treating tuberculosis. You might have explained that, but I missed it.
Laube:
This protein, lymphotactin, is key to forming the granuloma, and the authors felt that if they could suppress protein production, it might affect granuloma formation. In their murine model, the authors showed that lymphotactin production was suppressed with the interfering RNA, and there was an effect on the granuloma process. It wasn't forming as tightly. Suppression of lymphotactin also seemed to have unintended consequences because other pathways were also affected. For instance, the interferon γ response was affected and decreased as well, and T lymphocyte numbers were decreased with this suppression. So they think there's a whole milieu surrounding the granuloma formation process that could be affected when one pathway is targeted.
Hill:
This is what I'm missing. The idea is that if you have less granuloma formation, the organism would then be more accessible to chemotherapy. Is that the idea?
Laube:
No, they never said that.
Hill:
So why? I would think the way to treat tuberculosis would be with chemotherapy to eradicate the organism, and who cares about the granulomas? I'm missing something.
Laube:
They're trying to stop the process, and it doesn't look like chemotherapy works. For whatever reason—maybe because the drug doesn't get there properly, or there's not enough of it. Or they're thinking that if they target the various pathways, the disease process will be affected.
Hill:
Right, so that gets at the accessibility. If the organism is walled off inside the granulomas, chemotherapy agents can't get access. So perhaps that is the connection?
Laube:
Right, it may be. They did not discuss that, but it's a fair point. It may be that these interfering RNAs may be able to enter into these granuloma-type environments to a greater degree.
Footnotes
- Correspondence: Beth L Laube PhD, Johns Hopkins Children's Center, David M Rubenstein Child Health Building, 200 North Wolfe Street, Suite 3015, Baltimore, MD 21287-2533. E-mail: blaube{at}jhmi.edu.
Dr Laube presented a version of this paper at the 53rd Respiratory Care Journal Conference, “Aerosol Drug Delivery in Respiratory Care,” held June 6–7, 2014, in St Petersburg, Florida, and at the 9th International Symposium on Respiratory Diseases & ATS, held November 8–10, 2013, in Shanghai, China.
↵* James B Fink PhD RRT FAARC, James B Fink LLC, San Mateo, California, and Division of Respiratory Therapy, Georgia State University, Atlanta, Georgia, representing Aerogen.
↵† Neil R MacIntyre MD FAARC, Division of Pulmonary and Critical Care Medicine, Duke University, Durham, North Carolina, representing InspiRx.
↵‡ Jason A Suggett PhD MBA, Monaghan Medical.
- Copyright © 2015 by Daedalus Enterprises
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.
- 24.↵
- 25.
- 26.
- 27.
- 28.
- 29.
- 30.
- 31.
- 32.
- 33.
- 34.
- 35.
- 36.
- 37.
- 38.
- 39.↵
- 40.↵
- 41.
- 42.
- 43.
- 44.
- 45.
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.
- 52.
- 53.
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.
- 65.
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.
- 83.
- 84.
- 85.
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 113.↵
- 114.↵
- 115.↵
- 116.↵
- 117.↵
- 118.↵
- 119.↵
- 120.
- 121.
- 122.↵
- 123.↵
- 124.↵
- 125.↵
- 126.↵
- 127.↵
- 128.↵
- 129.↵
- 130.↵
- 131.↵
- 132.↵
- 133.↵
- 134.↵
- 135.
- 136.↵
- 137.↵
- 138.↵
- 139.↵
- 140.↵
- 141.↵





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}