

Abstract
Traumatic brain injury is a problem that affects millions of Americans yearly and for which there is no definitive treatment that improves outcome. Continuous brain tissue oxygen (PbtO2) monitoring is a complement to traditional brain monitoring techniques, such as intracranial pressure and cerebral perfusion pressure. PbtO2 monitoring has not yet become a clinical standard of care, due to several unresolved questions. In this review, we discuss the rationale and technology of PbtO2 monitoring. We review the literature, both historic and current, and show that continuous PbtO2 monitoring is feasible and useful in patient management. PbtO2 numbers reflect cerebral blood flow and oxygen diffusion. Thus, continuous monitoring of PbtO2 yields important information about both the brain and the lung. The preclinical and clinical studies demonstrating these findings are discussed. In this review, we demonstrate that patient management in a PbtO2-directed fashion is not the sole answer to the problem of treating traumatic brain injury but is an important adjunct to the armamentarium of multimodal neuromonitoring.
- Licox
- neurovent
- cerebral pressure autoregulation
- cerebral blood flow
- oxygen reactivity
- traumatic brain injury
- brain tissue oxygenation
Introduction
Traumatic brain injury (TBI) is a physical insult to the head that results in a clinically detectable alteration in cognitive processing that affects >2.5 million people per year in the United States and an estimated 10 million people worldwide.1–4 The cognitive dysfunction that results from TBI exists along a continuum with a subtle alteration in sensorium on the mild end and frank coma on the severe end. Despite decades of research into the pathophysiology of TBI, there is currently no reliable treatment option for TBI or its cognitive and psychological sequelae.
The underlying assumption of TBI research is that brain injury causes a pathological change in cerebral physiology that directly leads to a cascade of secondary injury. This secondary injury culminates in neuronal death, which can yield widespread symptoms, including cognitive dysfunction. Preventing secondary injury and neuronal death is challenging because TBI has been shown to cause a derangement in a wide range of neurophysiological parameters. It is the investigators' task to determine which of these parameters correlates most closely with the fundamental pathophysiology and can be used to monitor the extent of disease and the response of the brain to treatment.
Limitations to treating TBI are related to the information that can be gathered about the injured brain. Here, we review literature suggesting that one of the fundamental pathophysiological changes that occurs after TBI is a derangement of oxygen delivery to neural tissue. Practically, this postulate suggests that brain tissue oxygenation (PbtO2) should be monitored in severe cases of TBI and that maintaining a normal or elevated PbtO2 should improve outcomes after brain trauma. In this review, we first discuss the historical link between brain oxygenation and TBI. Then we focus on the different methods of measuring brain oxygenation. We then describe the ability of continuous brain tissue oxygenation monitoring to yield information about the cerebral autoregulation status of the patient. We explain the relationship between PbtO2 and lung function. Finally, we focus on the limitations of measuring brain tissue oxygenation and future directions in the field of multimodal monitoring for traumatic brain injury.
Why Monitor Brain Tissue Oxygen After Injury?
Operating under the assumption that alterations in brain physiology directly cause the dysfunctions that define TBI, it is important to monitor physiological parameters that correlate with disease severity. Indeed, enhanced monitoring is a hallmark of modern ICU care and has been shown to correlate with positive outcomes.5 Historically, pupil diameter, corneal reflexes, and other aspects of the neurological exam have been the key variables used by clinicians to monitor disease progression in TBI. These variables are deduced from the physical exam and are analogous to auscultating the heart and lungs during cardiopulmonary failure. The neurological examination remains the mainstay of ICU monitoring for patients with brain injury. Unlike the progress that has been made in cardiovascular and respiratory monitoring, in which multiple data points are available to help guide treatment, streamlined multimodal monitoring protocols for the brain are absent.
Outside of the neurological examination, the mostcommon physiological parameter monitored in TBI is intracranial pressure (ICP). ICP is conceptualized by the Monro-Kellie doctrine, which states that intracranial pressure is a function of the amount of brain tissue, blood, and cerebrospinal fluid present within the skull.6 From this general doctrine, ICP can be used to estimate the cerebral perfusion pressure and, hence, the amount of oxygen that is reaching the brain per unit of time. However, despite representing a major step forward in monitoring, ICP does not represent a complete picture of pathophysiology during TBI. Cerebral oxygenation, cerebral metabolism, cerebral blood flow, and autoregulation status are all useful adjuncts to the management of the brain-injured patient. ICP monitoring alone does not track the underlying pathophysiological processes that govern the degree of injury and potential for recovery after brain injury.
A key physiological variable in TBI is brain oxygenation. Exemplifying the tight relationship between brain injury and brain oxygenation, very early papers often categorized anoxic brain injury and TBI together as a single disease, given their similarities in clinical presentation.7 The importance of oxygen in TBI was only strengthened in the decades that followed,8–11 culminating in the seminal work by Chesnut et al,12 in which avoidance of secondary injury, primarily by maintaining oxygenation and blood pressure in the early stages after brain injury, correlated with positive outcomes. Thus, the amount of oxygen that the brain tissue receives is a fundamental physiological process that is disrupted by TBI. Monitors that directly measure this cerebral physiology should, in theory, track disease severity and serve as determinants as to when more invasive treatments are needed. In this context, ICP and cerebral perfusion pressure alone, although important monitoring variables, may not predict outcome because they are only indirect metrics of the physiological processes underlying TBI.
How Should Brain Tissue Oxygen Be Measured?
The argument in favor of measuring brain oxygenation is simple: By closely following and maintaining cerebral oxygenation, it may be possible to minimize the impact of secondary injury. However, it is not clear how brain oxygenation should be measured. Early studies measured the degree of hypoxia in the brain after injury by analyzing autopsy studies of brain-injured patients. They noted that areas of local and global ischemia that occurred after TBI correlated with disease severity and also that certain areas of the brain (eg, hippocampus) were disproportionately affected after injury.13,14 These studies showed that brain ischemia and TBI are inextricably linked, bolstering the view that brain oxygenation is the primary pathophysiological change in TBI.
After postmortem studies demonstrated that brain oxygen was a key cause of mortality, subsequent research focused on methods to measure oxygenation during the acute phase of the illness and increasing oxygen in the brain as much as possible. The first studies implemented peripheral oxygen saturation measurements as a proxy for brain tissue oxygen and mean arterial pressure for cerebral perfusion pressure.11,12,15 These studies were a landmark in the field of TBI and demonstrated that even moderate periods of hypoxia and hypotension were sufficient to cause a large increase in mortality after TBI. However, there are a number of problems with using peripheral physiological markers to monitor disease severity in TBI. First, because of cerebral autoregulation that maintains cerebral blood flow for varying degrees of ICP and mean arterial pressure, it is simply not possible to know the cerebral perfusion pressure without knowing the ICP. Second, hypoxia that is measured peripherally is affected by a number of factors outside of the brain, including the oxygen extraction by peripheral tissue. Thus, peripheral oxygen is a very coarse measure of brain oxygenation.
To overcome these issues, there needs to be a direct method of measuring brain oxygenation. Measuring cerebral blood flow is a strategy to obtain comparable information about the brain. This can be done directly using positron emission tomography16 or xenon computed tomography (CT).17,18 These methods provide accurate and useful measures of cerebral blood flow; however, these techniques only provide a snapshot in time and cannot be used to continuously monitor patients. Thus, positron emission tomography and xenon CT are imaging techniques that are largely used to measure cerebral perfusion dynamics after an ischemic stroke but have limited utility in the continuous measurement of brain oxygenation.
Another method of measuring brain oxygenation is by placing a monitor in the jugular bulb and quantifying the percentage saturation of the venous blood returning to the heart (SjvO2). SjvO2 is a global measure of how much oxygen is being extracted by the entire brain. SjvO2 desaturation, defined as a value of <50–55% for >10 min, has been associated with poor neurologic outcome.19–21 Conversely, an SjvO2 elevated >75% is also associated with poor outcome in patients with severe TBI.20 Due to the association between abnormal values and poor outcome, SjvO2 has been used as a primary outcome in clinical trials.22 However, SjvO2 is often subject to artifacts due to patient head position and the proximity of the probe to the jugular bulb2321,24,25 As a result, it has not been used widely in routine TBI critical care. To overcome the unreliable nature of the SjvO2 and to gain a better understanding of the oxygen delivery to neural tissue, electrodes were developed to directly measure the PO2 in brain tissue.
The Technology of the Partial Pressure of Brain Oxygen
There are 2 main methods to measure oxygen in the brain. The first is based on the Clark electrode, which is a general purpose electrode used to measure the PO2.26 The Clark electrode works by opposing 2 metallic surfaces (a gold cathode and a silver anode) in an aqueous electrolyte potassium chloride solution and allowing oxygen to diffuse into the solution. The oxygen carries forth an electrochemical reaction and creates an electric potential between the 2 surfaces, thus allowing the resulting current to be measured (Fig. 1A). The greater the amount of oxygen, the greater the electric current generated and, thus, the larger the reading of the Clark electrode. This electrode is used extensively in medicine to measure oxygen partial pressure in blood28 and muscle.29 The Licox PbtO2 monitoring system (Integra Life Sciences Corporation, Plainsboro, New Jersey) uses this same technology and applies it to neural tissue.30–32 The disadvantage of the Licox electrode is that it measures in a very focal space, thus limiting the capture of PbtO2 information to one area.33 Another disadvantage is that the amount of oxygen diffused in the electrolyte solution is dependent on the configuration of the anode and the cathode as well as the temperature of the surrounding tissue. Thus, each Licox electrode has its own temperature-current curve, which has to be calibrated individually for each patient. The newest versions of the Licox PbtO2 monitoring system come with a precalibrated card for each probe that allows the calibration information to be utilized without end-user calibration steps.
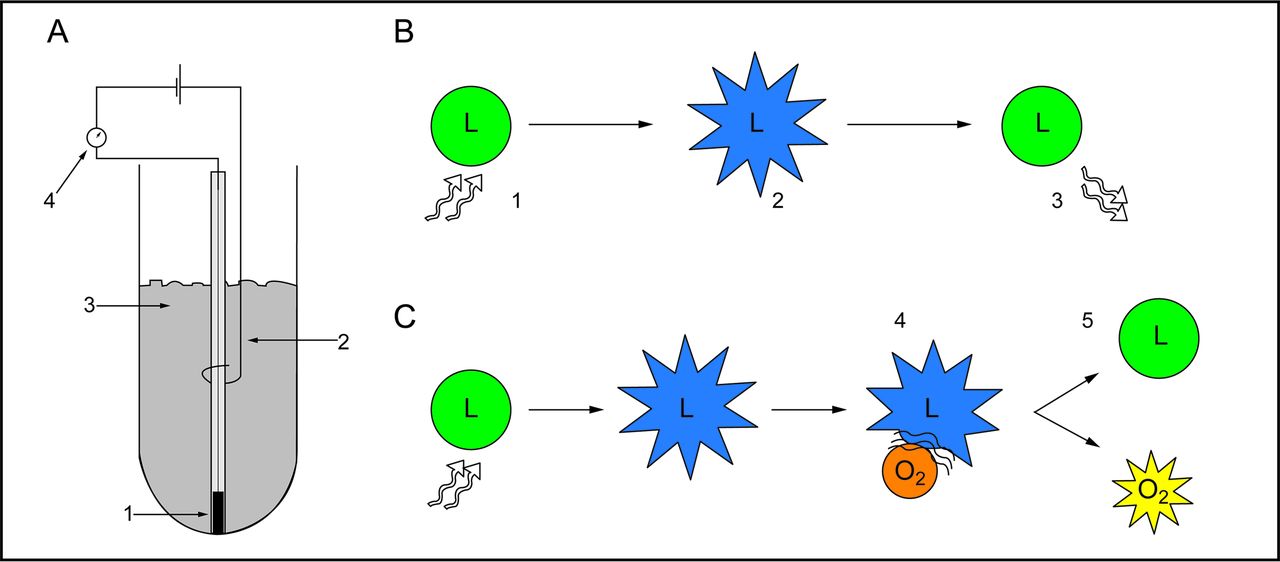
The technology of brain tissue oxygen monitoring. A: Illustration demonstrating the features of Clark electrode technology, including gold cathode (1), silver anode (2), and potassium chloride solution (3). Molecular oxygen is electrolytically reduced, which creates a current that is measured by the galvanometer (4). B: Schematic showing the concept of luminescence quenching. The properties of a luminophore (L) in the absence of oxygen are shown. Light is absorbed by the luminophore (1), which generates an excited state (2). The luminophore becomes deactivated and releases light (3), which can be measured. C: In the presence of molecular oxygen (O2), the excited luminophore collides with oxygen (4). This causes the luminophore to be deactivated without the emission of light (5). Adapted from Reference 27.
The second method of measuring PbtO2 uses fluorescence technology.34 These sensors contain a light source that shines on a medium containing a light-absorbing dye. This absorption processes is hindered by oxygen (Fig. 1B,C). With more oxygen in the medium, fewer photons are reabsorbed by the light detector and then converted into an electric current and amplified, yielding the PbtO2.
The initial fluorescence-based probe (Neurotrend) and the electrochemical-based probe (Licox) showed differences in threshold values in published clinical studies. Studies were done to assess whether differences in probe technology and design accounted for clinical differences and to assess whether performance in vivo was accurate as compared with in vitro conditions. Comparisons of the 2 probe types showed that the Neurotrend probe had a tendency toward higher PbtO2 values, which may have been related to differing positions of the oxygen sensor on the probe.35 The oxygen sensor on the Neurotrend probe was near the top of the probe, providing a closer proximity to gray matter after insertion. The Licox oxygen sensor, located at the tip of the probe, allows for consistent white matter measurements. Gray matter has higher PbtO2 measurement values than white matter, which could account for higher PbtO2 values when using Neurotrend. The sensing surface area of the Neurotrend probe was relatively small, thereby providing a more focal measurement of tissue oxygen tension. The Licox has a larger sensing surface area, which serves to average a greater volume of tissue and provide more consistent and reproducible measurements. Additionally, in vitro measurements showed that Licox was accurate with a range of 2.1–6.3% error, whereas Neurotrend had a percentage error that ranged from 2.9 to 7.4%, with the majority of this error seen when low oxygen tension was tested. In multiple in vitro tests, the electrochemical probe was found to be slightly more accurate, especially at low oxygen tension, which is important in distinguishing critical values in at-risk patients.30,35,36 In a study where Licox and Neurotrend catheters were placed in parallel, there was found to be a 6.25-mm Hg difference in PbtO2 readings between the probes, which could be clinically important in cases of low PbtO2.37 In addition, catheter malfunction was reported more frequently with the Neurotrend probe.37
Due to these findings, the Licox probe became the standard in the field. The Neurotrend went out of production in 2004. However, there are known limitations to using the Licox probe clinically. PbtO2 measurements can take up to 2 h to equilibrate in vivo. In vitro studies show that in conditions of 6% oxygen at 37°C, the Licox probe gives an accurate reading in <100 s. When used clinically, the probe first reads a PO2 consistent with atmospheric oxygen. Once the probe is inserted in the brain parenchyma, it slowly corrects to an accurate reading; however, this adaptation time averages 79 min (range 20–150 min). For practical use, this means that ≥1 h should pass before using any readings of the probe to assess the clinical situation.38 As mentioned above, positioning of the catheter in the white matter is optimal; however, malpositioning of the probe in an area of focal ischemia or hematoma can give misleading results.39 Thus, confirmation of probe placement by CT scan is standard.
Recently, a newer technology has emerged that utilizes the fiberoptic luminescence quenching properties to measure PbtO2 and simultaneously measures ICP and temperature (Neurovent-PTO, Raumedic, Mills River, North Carolina). This technology has been compared with Licox and shows results similar to the defunct Neurotrend probes. Again, with the Neurovent-PTO, higher PbtO2 values were noted, especially when testing in high PaO2 situations.27,40,41 Differences between the probes exist and have been partially attributed to the different sampling sizes of the probes.40,41 The Licox has a sampling size of 13 mm2, whereas the Neurovent-PTO samples 22 mm2 of the surrounding brain tissue. In vitro comparisons of these probes have demonstrated that, although both are accurate, Licox values more closely approximate the reference value when examining lower PbtO2,42 and Neurovent-PTO has a shorter response time and higher response to oxygen challenge.42,43 No probe has been demonstrated as superior, and both produce results within a clinical margin of era. To date, the majority of clinical studies and multi-center clinical trials have utilized the Clark electrode technology in the Licox probe.
Validation of Cerebral Oxygen Monitoring
With the technical challenge of measuring PbtO2 overcome, there remained the need to validate cerebral oxygen monitoring and show its clinical applicability. Clinically feasible continuous brain tissue oxygen monitoring should be safe and detect changes in PbtO2 in the setting of dynamic physiological parameters. Many preclinical studies were done in animal models before the routine clinical use of the technology. A study in the rat brain showed that contusion in the vicinity of the probe lowered the PbtO2 reading. In that study, van den Brink et al39 also demonstrated that a small zone of edema was present histologically in the region surrounding the probe, yet overall tissue damage related to the probe was minimal. In a series of normal cats, Zauner et al44 demonstrated a mean PbtO2 of 42 mm Hg, which decreased by 29% with hyperventilation. Manley et al45 showed that changes in brain tissue oxygen coincided with the physiological shifts that occur during hemorrhagic shock. Using a swine model, they demonstrated that a decrease in PbtO2 was seen with hemorrhage and recovered with resuscitation. Changes in ventilation provided an increase in PbtO2 in the setting of hypoventilation and a decrease in PbtO2 with hyperventilation.45 Hyperventilation exacerbated the decrease in PbtO2 during experimental hemorrhagic shock.46 These studies, while showing the feasibility of PbtO2 monitoring, also demonstrated the ill effects of hyperventilation, which had been used as a standard treatment for patients with increased intracranial pressure. Thus, PbtO2 monitoring began to show promise as an adjunct to current monitoring techniques in the setting of traumatic brain injury.
Early studies in patients focused on the clinical applicability of the PbtO2 technology. Because hypoxia after severe brain injury is a significant contributor to cell death, a threshold value that could guide patient treatment was sought. van Santbrink et al47 demonstrated in brain-injured subjects that having low brain tissue oxygen, as measured by the Licox probe, was correlated with an increased risk of death. This study was followed by a larger study involving 101 subjects that demonstrated that PbtO2 levels of <15, 10, and 5 mm Hg were all associated with increased risk of death or bad outcome.48 Valadka et al30 showed that prolonged PbtO2 of <6 mm Hg was not compatible with life and that PbtO2 <15 mm Hg for >30 min was associated with increased mortality.
Hyperventilation and Carbon Dioxide Reactivity
Early benefits of using continuous PbtO2 monitoring included discerning the relationship between hyperventilation and brain tissue oxygen. Hyperventilation induces hypocapnia, and PaCO2 is a potent cerebral vasomodulator. The arterial response to CO2 results in cerebral vasodilation during episodes of hypercapnia and vasoconstriction with hypocapnia.49 The vasoconstriction that occurs with hypocapnic hyperventilation subsequently decreases cerebral blood flow and cerebral blood volume.50–52 This was historically touted as a treatment for elevated ICP because of the direct correlation between ICP and cerebral blood flow/cerebral blood volume. A decrease in cerebral blood flow, as seen with hyperventilation, leads to a decrease in ICP. Despite the benefits of decreased ICP, the negative effect of hyperventilation has been demonstrated in a variety of settings. A randomized controlled trial compared the management of subjects with severe TBI using hyperventilation as a treatment modality. This trial showed worsening outcomes in the hyperventilation group at 3 and 6 months; however, the mechanism underlying the poor performance in the hyperventilation group was unexplained.53
Studies that examined cerebral blood flow by either xenon CT54 or positron emission tomography55 demonstrated a decrease in cerebral blood flow below ischemic thresholds with hyperventilation therapy. Eighty-four percent of subjects given a hyperventilation challenge demonstrated a substantial decline in PbtO2 even when the overall reduction in PaCO2 was by only 2 mm Hg.56 This suggested that decrements in cerebral blood flow might lead to changes in PbtO2, which may underlie the ill effects of hyperventilation therapy.
Severely brain-injured patients often have spontaneous episodes of hyperventilation when managed on ventilator settings allowing spontaneous breaths. In these instances of hypocapnic hyperventilation, Carrera et al57 demonstrated that the decrease in PaCO2 still correlates with a decrease in PbtO2. This emphasizes that the PbtO2 response to decreased PaCO2 is not a function of the artificial nature of hyperventilation therapy. Attempts to modulate hyperventilation therapy by targeting SjvO2 revealed that maintaining a normal SjvO2 did not protect the PbtO2.23 This study, along with others,58 demonstrated that global measurements of brain oxygenation, such as with SjvO2, gives information complementary, but not identical, to that from regional PbtO2 measurements. It was also further confirmed that moderate hyperventilation decreases cerebral blood flow to a level that causes a decline in regional brain tissue oxygenation.
Brain Tissue Oxygen and Cerebral Pressure Autoregulation
Research suggested a positive correlation between cerebral blood flow and PbtO2.59–61 This led to the question of whether continuous PbtO2 monitoring could serve as a surrogate for cerebral blood flow and hence deliver information about the cerebral autoregulation status of the patient. Knowledge of the cerebral autoregulation status of a patient with a severe head injury can help to guide treatment and determine outcomes. Cerebral pressure autoregulation is based on the notion that cerebral vessels respond to changes in blood pressure by dilation and constriction as appropriate, similar to the CO2 reactivity explained above. This locally mediated change in vessel caliber allows cerebral blood flow to be maintained over a wide range of mean arterial pressures before the system can no longer compensate.62,63 In brain-injured patients, there is often a loss of cerebral autoregulation allowing cerebral blood flow decreases in the face of decreasing blood pressure.64
Studies of PbtO2 showed trends in physiological factors suggesting that PbtO2 provides information about cerebral blood flow. However, the majority of studies evaluating brain tissue oxygen were performed in experimental injury models or subjects with severe head injury; hence, the results determining the influence of normal cerebral physiology on PbtO2 were indeterminate. A study in uninjured swine evaluated normal cerebral physiology by monitoring PbtO2 in the setting of various challenges. In this study, it was confirmed that PbtO2 increased linearly with increased end-tidal CO2 (PETCO2) yet remained constant over a wide range of mean arterial pressures.60 In another study, comparisons between uninjured animals and subjects with severe TBI demonstrated that uninjured animals showed evidence of autoregulation, whereas injured subjects showed tight linear correlations between cerebral perfusion pressure and PbtO2.65 Direct comparisons of cerebral blood flow and PbtO2 confirmed a tight linear correlation. Using xenon CT, a correlation between PbtO2 and both regional and global cerebral blood flow was demonstrated in injured patients.59,61 Later, Jaeger et al66 used the continuous cerebral blood flow probe (Bowman Perfusion Monitor, Hemedex, Cambridge, Massachusetts) in combination with Licox to further verify this relationship. They demonstrated a statistically significant Pearson correlation coefficient of ≥0.6 between cerebral blood flow and PbtO2 in the majority of subject intervals examined. Thus, both PbtO2 and cerebral blood flow are able to demonstrate cerebral autoregulation over a range of blood pressures. Figure 2 illustrates the similarities in the pressure response curves for PbtO2 and cerebral blood flow and the correlation between the 2 physiological measures.

Cerebral autoregulation and the relationship between cerebral blood flow and brain tissue oxygen (PbtO2). A: PbtO2 remains stable over a range of mean PaO2 from approximately 50 to 150 mm Hg. B: Along this same range of mean PaO2 values, cerebral blood flow also remains stable in a subject that shows appropriate cerebral autoregulation. C: Schematic demonstrating that PbtO2 and cerebral blood flow are linearly related.
Changing PbtO2 levels replicate the changes seen in cerebral blood flow with blood pressure challenges, whereby PbtO2 and cerebral blood flow are correlated. Menzel et al65 subsequently introduced the cerebral perfusion oxygen reactivity index to follow autoregulation status. This index, which represents the percentage change in PbtO2 divided by the percentage change in cerebral perfusion pressure, was found to be a value of <1 in physiologic conditions of uninjured brain. In injured brain with loss of autoregulation, the cerebral perfusion oxygen reactivity was >1. Lang et al67 took a similar analytic approach. They looked at the interaction between blood pressure changes and PbtO2 among 14 injured subjects and demonstrated that subjects with intact autoregulation demonstrated smaller changes in PbtO2 with cerebral perfusion pressure changes.67 The standard approach to assess cerebral autoregulation is to calculate a cerebrovascular pressure reactivity index. This index evaluates the response of ICP to changes in mean arterial pressure. Jaeger et al68 used this standard measure of autoregulation and compared it with a brain tissue oxygen pressure reactivity index and found the 2 measures to be highly correlated. This measure of autoregulation appears to be robust, since a study in pigs comparing the Licox probe to the Neurovent-PTO probe demonstrated that the brain tissue oxygen pressure reactivity was measurable and consistent between the 2 probes.69
Cerebral Oxygen Reactivity
The close relationship of PbtO2 to PaO2 is easily demonstrated in studies performing oxygen challenges to maintain normobaric hyperoxia. Testing the functionality of a PbtO2 parenchymal probe is routinely done with such an oxygen challenge. In this challenge, FIO2 is increased to 1.0, and a change in PbtO2 is observed. Due to the universality of an increase in FIO2 leading to an increase in PbtO2, the absence of an increase in PbtO2 with an oxygen challenge represents a faulty or malpositioned probe.
Although normobaric hyperoxia universally causes an increase in PbtO2, the character of the increase can vary. Patients with low cerebral blood flow and low PbtO2 baseline values show a smaller increase in PbtO2 with hyperoxia.70,71 This varying degree of reactivity to an oxygen challenge has been defined as relative tissue oxygen reactivity.47,72 Tissue oxygen reactivity is defined as the change in PbtO2 divided by the change in PaO2, divided by the baseline PbtO2 (tissue oxygen reactivity = [ΔPbtO2/ΔPaO2]/PbtO2 baseline). This relative measure gives an important way to process the oxygen challenge information. For any given oxygen challenge in which the FIO2 is increased to 1.0, there can be a varying degree of change in PaO2. Some of this variability is related to the starting FIO2 and starting PaO2 at the beginning of the challenge. Additionally, the change in PbtO2 can vary based on the starting PbtO2. Therefore, the relative tissue oxygen reactivity gives an accurate way to compare responses across different conditions.
Initial studies using this metric demonstrated that subjects with a lower relative tissue oxygen reactivity had a better outcome.47 This implied that an increased reactivity to an oxygen challenge represented a disturbed autoregulation for oxygen. Additional studies confirmed these results in a larger sample size and demonstrated that an increased tissue oxygen reactivity within the first 24 h of injury was significantly associated with a poor outcome.72 Subjects with a favorable outcome, as defined by the Glasgow Outcome Score, had a mean relative tissue oxygen reactivity of 0.61, whereas subjects with unfavorable outcome had a mean relative tissue oxygen reactivity of 1.03. van Santbrink et al72 proceeded to illustrate 3 patterns of PbtO2 response to hyperoxia. Type A shows a sharp increase of PbtO2 that reaches a plateau within minutes, when FIO2 is increased. An FIO2 challenge that results in a sharp increase in PbtO2 followed by a gradual increase that continues without a plateau within 15 min is labeled Type B. A Type C response is a hybrid in which a sharp increase initially plateaus but then results in a second breakthrough increase in PbtO2. It was observed that Type A and B patterns occurred more frequently (40 and 44%, respectively), and there was a trend toward improved outcome in subjects showing Type A curves (P = .06). Given the importance of the change in PaO2 to relative tissue oxygen reactivity, it is unlikely that cerebral factors affect oxygen reactivity in isolation.
Brain Tissue Oxygen and Oxygen Diffusion
The importance of PbtO2 in relation to cerebral blood flow, PaO2, cerebral pressure autoregulation, CO2 reactivity, and O2 reactivity should not be overlooked. However, the assumption that low PbtO2 also represents cerebral ischemia is simplistic and inaccurate. Ischemia, the balance between oxygen delivery and oxygen metabolism, is an elusive target. According to the Fick principle, the amount of oxygen that diffuses across the blood-brain barrier to the brain equals the cerebral blood flow times the difference in arterial and venous oxygen content. This is equivalent to the cerebral metabolic rate of oxygen plus the rate of accumulation of oxygen in the tissue. This equation can be rearranged to show that the cerebral metabolic rate of oxygen is nearly equivalent to the cerebral blood flow times the O2 off-loaded from hemoglobin plus the cerebral blood flow times the O2 dissolved in plasma (Fig. 3A). Rosenthal et al73 studied injured subjects with PbtO2 and cerebral blood flow probes and demonstrated with multivariable analysis that PbtO2 is most dependent on the cerebral blood flow times the difference in arterial and venous oxygen tension. This corroborates the finding that PbtO2 is linearly related to PaO2 and cerebral blood flow.60 Thus, PbtO2 monitoring is a reflection of the dissolved oxygen within the plasma that diffuses across the blood-brain barrier rather than entire oxygen content or cerebral metabolism. Thus, PbtO2 is not an ischemia monitor, but low values can provide information about low PaO2 or cerebral blood flow. This also suggests that PbtO2 is not merely a surrogate for cerebral blood flow and that factors that influence the amount of dissolved plasma oxygen (such as pH, temperature, altitude, PaCO2, and allosteric effectors of hemoglobin) probably influence tissue oxygen reactivity.
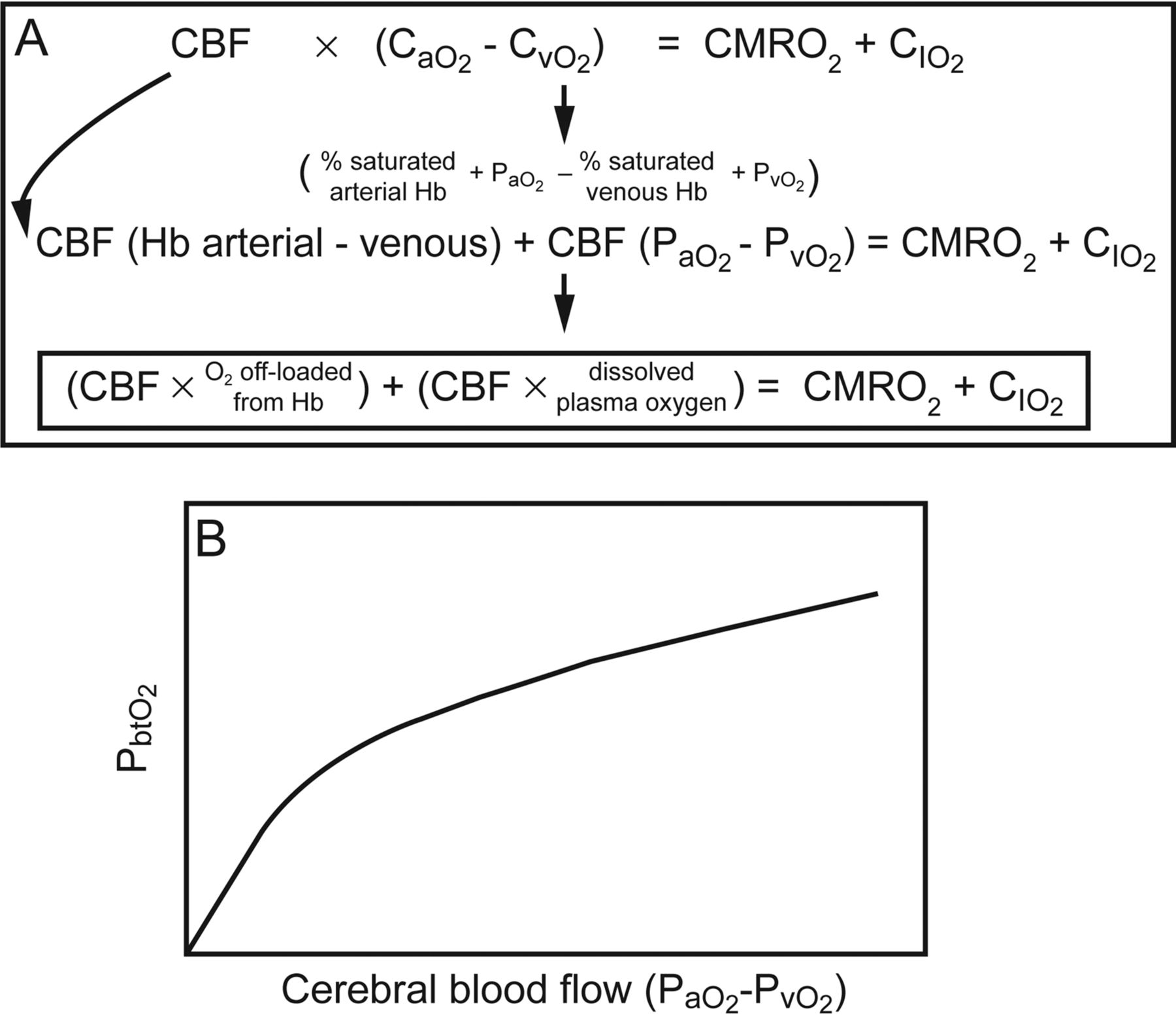
A: The Fick equation of cerebral oxygen metabolism can be rearranged and represented as denoted. B: Experimental data and multivariable analysis show that the brain tissue oxygen value (PbtO2) is most closely related to the product of cerebral blood flow (CBF) and the difference in dissolved plasma oxygen. CaO2 = arterial oxygen content; CvO2 = venous oxygen content; CMRO2 = cerebral metabolic rate of oxygen; CIO2 = rate of accumulation of oxygen in the tissue. Data from Reference 73.
Brain Tissue Oxygenation and the Role of the Lung
The strong influence of PaO2 on PbtO2 suggests that factors contributing to low PaO2 can affect PbtO2. One such factor in a mechanically ventilated patient is an inadequate FIO2. Although an attractive solution to correct a low PbtO2 in a ventilated patient is to increase the FIO2 and subsequently the PaO2, there are risks to prolonged normobaric hyperoxia. Prolonged FIO2 >0.6 is known to cause hyperoxic acute lung injury due to the production of reactive oxygen species and the cellular damage incurred on lung tissue.74,75 In brain tissue, hyperoxia can similarly cause cellular dysfunction and exacerbate brain injury. In studies of stroke and traumatic brain injury, although evidence exists that normobaric hyperoxia may be neuroprotective,76–79 there is counterevidence that hyperoxia does not improve outcome and is detrimental.77,79–82 Prolonged hyperoxia may provide some temporary benefits, such as a decrease in cerebral edema; however, this “benefit” is probably due to a compensatory cerebral vasoconstriction, which risks a decrease in cerebral blood flow and increased ischemia in vulnerable tissue.
In addition to these risks of hyperoxia, a solely FIO2-directed strategy to address a low PbtO2 may be a solution for the number but not for the cause of the problem. In a study examining oxygen reactivity in the context of lung injury, divergent patterns of oxygen reactivity were seen.83 Rosenthal et al83 examined PbtO2 responses to oxygen challenge while noting the lung function of the subject by assessing the PaO2/FIO2. Using the criterion that PaO2/FIO2 <250 mm Hg represents poor lung function (atelectasis, pneumonia, lung injury), responses to hyperoxia were compared in subjects with PaO2/FIO2 >250 mm Hg versus PaO2/FIO2 <250 mm Hg. As expected, both groups showed consistent correlations of increased PbtO2 with increased PaO2. However, the pattern of increase differed. Brain-injured subjects with normal lung function showed a sharp increase in PbtO2 that reached a plateau quickly, similar to van Santbrink Type A oxygen reactivity (Fig. 4A). Subjects with lung injury showed a slower response to PaO2 increase that did not plateau immediately, as is seen in van Santbrink Type B (Fig. 4B). Rosenthal et al73 did not find a relationship between tissue oxygen reactivity and outcomes, possibly due to sample size. However, when the tissue oxygen reactivity is calculated based on the published data, tissue oxygen reactivity is lower in an example with PaO2/FIO2 >250 mm Hg and higher in an example with PaO2/FIO2 <250 mm Hg. Thus, oxygen reactivity detected during an oxygen challenge may indicate as much about the injury status of the lung as it does the brain.

Effect of lung function on brain tissue oxygen in the presence of good lung function (PaO2/FIO2 >250). The brain tissue oxygen (PbtO2) increases sharply and plateaus in the presence of an oxygen challenge (A). In the setting of lung injury (PaO2/FIO2 <250) the response to hyperoxia is an increase in PbtO2 that is slow and of lower amplitude (B). Data from Reference 73.
Decreased PbtO2 can be a signal of poor pulmonary gas exchange. Because PbtO2 is so closely linked to PaO2, changes in FIO2 are directly reflected in changes in PbtO2. In fact, the opposite is also true, in that a spontaneous decrease in PbtO2 can often represent poor pulmonary oxygenation and a low PaO2. In PbtO2-mediated treatment, the first step in treating a low PbtO2 involves verifying that PaO2 is ≥100 mm Hg. Changes in lung function, such as atelectasis, a new pneumonia, or developing ARDS, will cause a decrease in PaO2 to <100 mm Hg, which is often first detected by a drop in continuous PbtO2 measurements.
Lung-Protective Strategies and Brain Tissue Oxygen
Because the injury status of the lung influences the PbtO2 response, preventing lung injury may be an important adjuvant to brain injury treatment. However, there has been hesitancy to adopt the accepted lung-protective strategies in traumatic brain injury. ARDS is frequently seen in the setting of trauma; patients with concomitant ARDS and TBI are not uncommon. Additionally, the need for mechanical ventilation, as is universally the case in patients with severe TBI, increases the risk for ventilator-induced lung injury. The ARDS Network protocol (ARDSNet) has become the standard of care for preventing and treating ARDS. This protocol involves a lower tidal volume (6 mL/kg) and higher PEEP. This protocol has been shown to reduce mortality in a large multi-center randomized controlled trial.84 However, in this trial and in other related trials exploring lung-protective strategies, patients with brain injury have been excluded. The exclusion of patients with brain injury is due to the concern that an increased PEEP will increase ICP. A few studies have looked at this in animal and the results suggest that an increased PEEP can be safely administered as long as the PEEP does not exceed the ICP.85–87
However, the interaction between lung-protective strategies and brain tissue oxygen has not been thoroughly explored. As the above-mentioned paper by Rosenthal et al83 suggests, poor lung function is bad for brain tissue oxygen. This has been demonstrated by another group that examined 78 subjects with severe TBI and found a correlation between PaO2/FIO2 and PbtO2.88 Oddo et al88 showed that poor PaO2/FIO2 was an independent risk factor for poor PbtO2, thus concluding, along with Rosenthal et al,83 that a lung-protective strategy is good for the brain.
Animal studies have demonstrated that a low tidal volume lung-protective strategy can be safe and improve PbtO2. Bickenbach et al89 showed in a pig model of experimental ARDS that animals treated with low tidal volume ventilation (as compared with high volume) had higher PbtO2 and lower cerebral lactate levels. In a swine model of combined ARDS and TBI, Davies et al90 compared ARDSNet protocol with airway pressure release ventilation. Although PbtO2 was not directly measured, the ARDSNet group showed a better improvement in PaO2/FIO2 and fewer markers of cerebral injury as monitored by microdialysis.
Extreme lung-protective strategies, such as prone position, are a challenge in brain-injured patients. Prone position has been demonstrated to proffer a mortality benefit in patients with severe ARDS.91–93 The limited numbers of studies that have examined the efficacy of prone position in subjects with brain injury have found that prone position causes a slight increase in ICP; however, this is eclipsed by the clear benefit of improved oxygenation.94–98 One study evaluated PbtO2 in subjects with ARDS and concurrent subarachnoid hemorrhage and showed that prone position was well tolerated and resulted in significant increases in PbtO2.94 Although more studies examining the intersection of brain and lung need to be done, the available data suggest that if a ventilatory strategy results in an improved PaO2/FIO2 ratio, the benefits of improved PaO2 will be reflected in brain tissue oxygen monitoring and improved patient outcome.
Limitations
Brain tissue oxygen monitoring has become an important component of treatment in severe traumatic brain injury. However, standardized guidelines for routine implementation of PbtO2-directed therapy do not exist. The lack of standardized treatment guidelines is primarily due to the insufficient evidence that PbtO2-directed management improves outcomes better than ICP and cerebral perfusion pressure-directed treatment alone. In observational studies comparing historical cohorts with PbtO2-managed subjects, data show mortality and functional outcome benefits.99–101 However, many single-center studies have not demonstrated this benefit. In a study with 93 subjects, Meixensberger et al102 compared PbtO2-directed therapy with cerebral perfusion pressure-directed therapy and found no difference between groups. A larger study encompassing 629 subjects found no reduction in mortality rate with a PbtO2-guided treatment and additionally found worse functional outcome and increased utilization of hospital resources, yet the group with PbtO2 management had a higher overall injury severity at baseline than the ICP-only managed cohort.103 The trend of lack of benefit with PbtO2 management has been seen in many trials; however, the absence of standardized protocols to manage low PbtO2 limits the interpretation of these studies.33,104 As has been demonstrated, there are many factors that affect PbtO2. A lack of protocolized treatment strategy to improve PbtO2 numbers may have played a role in the failure of trials.105,106
There are some technical limitations related to PbtO2 management. Initially, the calibration of the device led to frequent errors and inconsistencies; however, the newer PbtO2 probes and monitoring devices have obviated that problem. The positioning of the probe can give misleading results. The probe should reside in white matter and ideally not be positioned in a focus of injured brain. Although positioning of the probe in injured brain yields important information about the injured tissue, it gives limited information as to the oxygen status of the surrounding tissue that may be vulnerable yet recoverable. Placement of a PbtO2 probe within or in close proximity of a contusion yields lower values.107 Thus, PbtO2 is a regional measure, and translating changes from a regional probe into conclusions about the global state of the brain has its own set of limitations.
Initial studies using PbtO2 hoped for a continuous measure of cerebral metabolism and an indication of cerebral ischemia. However, as has been demonstrated, reduced PbtO2 indicates low PaO2 or low cerebral blood flow, not total oxygen content or cerebral metabolism.73 In fact, in can be argued that the most important information derived from continuous PbtO2 data is the interface between the lung and the brain.
Future Directions
It has been recognized that continuous monitoring of PbtO2 has the ability to influence treatment of traumatic brain injury. PbtO2 monitoring has been shown to be feasible, and it has been demonstrated that injured patients have episodes where PbtO2 is abnormally low, despite normal ICPs. Despite the numerous single-center trials that have been done, a large randomized controlled trial is needed to effect standardized changes in the treatment guidelines.32,108 A phase 2 randomized clinical trial of the safety and efficacy of PbtO2 monitoring in the management of severe TBI (BOOST 2) has been completed and demonstrates that an ICP plus PbtO2-directed treatment strategy is feasible and safe.109,110 Definitive studies are under way to demonstrate whether PbtO2-directed therapy is superior to ICP-directed treatment and leads to better outcomes. Additionally, new technologies in the form of noninvasive infrared spectroscopy measurements of cerebral oxygenation show promise.111,112
Summary
In summary, technological advances have made continuous PbtO2 monitoring possible, and studies incorporating PbtO2 have demonstrated that subjects with low values do poorly. There is a tight relationship between PbtO2, cerebral blood flow, and PaO2, which underscores the important interaction between the lungs and the brain. Limitations to the universal utilization of PbtO2 technology involve the invasive nature of the monitoring and the lack of standardized guidelines. Thus, the future of PbtO2 monitoring includes noninvasive monitoring techniques and the creation of formal PbtO2-directed treatment recommendations.
There is no one number that can be used to treat the brain, just as no single number is used to treat the heart. Directed interventions for brain injury require a deep understanding of cerebral physiology and the tools to acquire and visualize the data. Continuous brain tissue oxygen monitoring is not the single answer to all of the problems involved with the management of patients with TBI. However, PbtO2 monitoring adds another data point that can be utilized to facilitate treatment goals.
Footnotes
- Correspondence: Geoffrey T Manley MD PhD, University of California, San Francisco, 1001 Potrero Avenue, Building 1, Room 101, San Francisco, CA 94110. E-mail: manleyg{at}ucsf.edu.
The authors have disclosed no conflicts of interest.
- Copyright © 2016 by Daedalus Enterprises
References
- 1.↵
- 2.
- 3.
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.
- 10.
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.
- 79.↵
- 80.
- 81.
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.
- 93.↵
- 94.↵
- 95.
- 96.
- 97.
- 98.↵
- 99.↵
- 100.
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Introduction
- Why Monitor Brain Tissue Oxygen After Injury?
- How Should Brain Tissue Oxygen Be Measured?
- The Technology of the Partial Pressure of Brain Oxygen
- Validation of Cerebral Oxygen Monitoring
- Hyperventilation and Carbon Dioxide Reactivity
- Brain Tissue Oxygen and Cerebral Pressure Autoregulation
- Cerebral Oxygen Reactivity
- Brain Tissue Oxygen and Oxygen Diffusion
- Brain Tissue Oxygenation and the Role of the Lung
- Lung-Protective Strategies and Brain Tissue Oxygen
- Limitations
- Future Directions
- Summary
- Footnotes
- References
- Figures & Data
- Info & Metrics
- References