

Abstract
Infection with SARS-CoV-2 in select individuals results in viral sepsis, pneumonia, and hypoxemic respiratory failure, collectively known as COVID-19. In the early months of the pandemic, the combination of novel disease presentation, enormous surges of critically ill patients, and severity of illness lent to early observations and pronouncements regarding COVID-19 that could not be scientifically validated owing to crisis circumstances. One of these was a phenomenon referred to as “happy hypoxia.” Widely discussed in the lay press, it was thought to represent a novel and perplexing phenomenon: severe hypoxemia coupled with the absence of respiratory distress and dyspnea. Silent hypoxemia is the preferred term describing an apparent lack of distress in the presence of hypoxemia. However, the phenomenon is well known among respiratory physiologists as hypoxic ventilatory decline. Silent hypoxemia can be explained by physiologic mechanisms governing the control of breathing, breathing perception, and cardiovascular compensation. This narrative review examines silent hypoxemia during COVID-19 as well as hypotheses that viral infection of the central and peripheral nervous system may be implicated. Moreover, the credulous embrace of happy hypoxia and the novel hypotheses proposed to explain it has exposed significant misunderstandings among clinicians regarding the physiologic mechanisms governing both the control of breathing and the modulation of breathing sensations. Therefore, a substantial focus of this paper is to provide an in-depth review of these topics.
Introduction
“When faced with doubtful situations, we are likely to jump to new and attractive conclusions, because of ease of recall. Ironically, it is in these circumstances that our clinical judgement is most often needed.”
Dipit Sahu1
During the first year of the COVID-19 pandemic, reports emerged in the medical literature describing what many authors believed was a uniquely COVID-19-related phenomenon of silent, happy, or apathetic hypoxia.2-7 Because hypoxia occurs at the tissue level, it is hypoxemia that is actually being described. To avoid confusion in this presentation, the term hypoxia will be used either in its correct connotation or when citing other sources directly.
The alleged uniqueness of silent hypoxemia, amid the confusion and apprehension of a terrifying pandemic, all but guaranteed its rapid dissemination across social and mainstream media platforms. By July 2020, there were only 4 relevant COVID-19 publications listed on PubMed (of which only 2 were research articles), whereas a Google search using the term “happy hypoxia” produced over 2 million results.1 The trivializing descriptor happy has been replaced with the preferred term silent hypoxemia.8,9
Initial reports from China mentioned apparent silent hypoxemia only passingly.10,11 One noted that chest computed tomography findings of diffuse, severe lung injury were associated with relatively mild clinical presentations in some patients without complaint of dyspnea or signs of respiratory distress.11 The first published case report of silent hypoxemia described a man in his 60s who, despite presenting with cyanosis and a pulse oximetry saturation 66%, was calm and cooperative with barely audible breathing. However, with ambulation he exhibited pronounced tachypnea that quickly rose to 48 breaths/min.7 Upon hospital admission, his chest radiograph revealed diffuse bilateral lung opacites.
Other case reports followed describing stable patients with silent hypoxemia, some rapidly developed cardiovascular instability resulting in sudden death, whereas others recovered rapidly.3,5,6,11-14 As of this writing, only 2 relatively large case-control studies have been published.15,16 Given the backdrop of overwhelmed hospitals, it is understandable that concern over silent hypoxemia quickly evolved from curious novelty into alarm disproportionate to its apparent prevalence.
When reading these reports, one immediately notices that, when discussed at all, the neurophysiology of silent hypoxemia focuses entirely on intriguing hypotheses regarding COVID-19-related neurologic dysfunction. Conspicuously absent was any acknowledgment of more than a half century’s worth of physiological research into respiratory drive and dyspnea.17-21 A broader perspective would have tempered the credulous embrace of novel hypotheses.
Only 2 early reports referenced studies describing the effects of hypobaric hypoxemia on respiratory drive and dyspnea or provided even a cursory overview of various mechanisms underlying dyspnea.7,14 In the 2 years that have passed since the onset of the COVID-19 pandemic, more literature has emerged examining the phenomenon of silent hypoxemia, placing it within the context of our established understanding of respiratory drive and dyspnea. The objective of this narrative review is to explore each of these topics as well as provide a critical analysis of early reports that animated the discussion surrounding silent hypoxemia.
Control of Breathing and Hypoxemic Cardiovascular Compensation
“There is no single mechanism that can be said to control ventilation. Many different mechanisms can be shown to be able to exert an influence on breathing under particular circumstances, although not all are in play at any one time.”
J.F. Nunn22
The enormous complexity in the neural control of breathing is beyond the scope of this paper. Only a cursory overview is provided here to facilitate the reader’s understanding of the ventilatory response during acute respiratory failure in general and COVID-19 in particular. Neurons residing in the central nervous system that regulate breathing are widely dispersed in the cerebral cortex, the hypothalamus, and other structures of the limbic/paralimbic system as well as the pons, medulla, and spinal respiratory neurons.
Basic rhythmicity is controlled by the central respiratory pattern generator, a column of neurons extending from the pons to the medulla (Fig. 1).23 It emanates in small regions of the ventrolateral medulla (pre–Bötzinger complex, lateral parafacial region, and post-inspiratory complex) with additional phase control shaped by structures in the pons (Kölliker-fuse nucleus, parabrachial complex).24 Together, these neural circuits influence breathing patterns under both physiologic and pathophysiologic conditions.25 Additional inputs to rhythmicity are generated in upper cervical inspiratory neurons (at the level of C-1 and C-2) that project into areas near the phrenic and intercostal motor neurons.25

Central nervous system illustrating the primary structures governing/modifying both the control of breathing and the perception of dyspnea. See text for descriptions. E-RDS = efferent-reafferent dissociation signaling (explaining dyspnea); KFN = Kölliker-fuse nucleus; NTS = nucleus tractus solitarius; PBC = parabrachial complex; P-BC= Pre-Bötzinger complex.
In addition, breathing rhythmicity is altered by cognitive, sensory, and emotional factors (eg, stress, pain, fear, anxiety) through activation of the limbic/paralimbic system.26,27,28 Volitional control of breathing (eg, talking, singing) is controlled by the motor cortex. Furthermore, emerging evidence suggests severe physical exertion directly enhances hyperpnea by stimulating mechanically and metabolically sensitive afferent nerve fibers in the skeletal muscles.29 These in turn projecting up into the medullary respiratory centers.
Chemosensitive structures in the ventral surface of the medulla regulate ventilation based upon local PaCO2 tension and its corresponding impact upon intracellular hydrogen ion concentration ([H+]). Additional afferent information arises from stimulation of peripheral chemoreceptors for PaCO2 in the carotid bodies that in turn enhance and quicken the response time of medullary chemoreceptors.30 Concomitant hypoxemia detected in the carotid bodies also increases the sensitivity of neighboring PaCO2 chemoreceptors (Fig. 2).31

Location of peripheral chemical and mechanical sensory receptors influencing the control of breathing and the sensation of dyspnea. Peripheral chemoreceptors located in the aortic arch and the carotid arteries are sensitive to both arterial CO2 and oxygen tension. Chest wall mechanoreceptors located at the origins and insertions of the ribs provide information on displacement, whereas muscle tendons provide information regarding tension development, and muscle spindles provide integrated information. Pulmonary receptors include irritant receptors in the central and peripheral airways (C-fibers), J receptors, and stretch receptors located in the alveolar walls. From Reference 68.
Other vagal afferent stimuli that modify breathing (through integration of proprioceptive information) include muscle spindle fibers, tendon organ receptors, and joint receptors in the chest wall32,33 as well as slow-adapting stretch receptors and irritant C-fibers in the airways and lung parenchyma (Fig. 2).29,34,35,36,37
Furthermore, acute lung injury results in the expression of pro-inflammatory cytokines in the brain stem that in turn induces tachypnea, even in the absence of chemosensitive and other afferent input.38 And although arterial baroreceptors in the carotid sinus and aortic arch primarily are involved in circulatory regulation, they also respond to hypotension by inducing hyperventilation.22
The Hypoxic Ventilatory Response
“Humans have been dealing with hypoxia in many different ways across evolution, both as breath-hold divers and as Indigenous permanent inhabitants of moderate to high altitude regions . . . proving that humans can thrive even in conditions of hypoxia.”
Eric Mulder39
Hypoxemia stimulates chemoreceptors in the carotid bodies that in turn modulate respiratory drive in a nonlinear fashion. The response is directly stimulated by decreased PaO2 tension or hypotension (“stagnant hypoxia”), not by SaO2.40 And in contrast to the stimulatory effects of acute hypercapnia on central respiratory drive, brain stem hypoxia exerts a directly depressant effect.22
The ventilatory threshold to hypoxemia occurs when PaO2 sensed in the carotid bodies is ≤ 60 mm Hg.40-42 The responses to acute hypoxemia (referred to as hypoxic ventilatory response) include tachypnea, hyperventilation, tachycardia, and elevated cardiac output, all of which rise proportionally as the severity of hypoxemia increases from mild to profound.42-45
Furthermore, the magnitude of the hypoxic ventilatory response is mediated by the corresponding PaCO2. When hypoxemia is induced in normal subjects, reducing PETCO2 tension to < 29 mm Hg prevents the hypoxic ventilatory response even when SpO2 is ≤ 70%. In contrast, the hypoxemic ventilatory response is induced systematically when PETCO2 is ≥ 34 mm Hg.46 That PETCO2 is normally ≤ 5 mm Hg below arterial values suggests that corresponding PaCO2 levels of ∼35 and ∼40 mm Hg, respectively, either suppress or facilitate the hypoxic ventilatory response. Historically, acute acclimatization hypoxic drive becomes the primary driver of ventilation only at an altitude of ∼13,000 ft when PaO2 reaches ∼45 mm Hg. Initial compensatory hyperventilation is mild, resulting in a PaCO2 ∼35–38 mm Hg. Over time PaCO2 stabilizes at ∼30 mm Hg, causing PaO2 to stabilize at ∼55 mm Hg.22 A more recent study in experienced mountaineers produced similar data (pH 7.44 ± 0.04, PaCO2 35 ± 5 mm Hg, PaO2 47 ± 8 mm Hg, SaO2 83 ± 5%) without discernable alterations in breathing frequency, heart rate, and blood pressure.47
Cardiovascular Responses to Worsening Hypoxemia
Worsening hypoxemia proportionally increases the cardiovascular response. During mild hypoxemia (PaO2 50–60 mm Hg, SaO2 85–90%), both young and older adults as well as those with cardiovascular disease respond with increased heart rate and cardiac output (Fig. 3).43 The ventilatory response appears mild until PaO2 reaches ∼50 mm Hg, and the initial minute ventilation (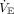 ) response usually stabilizes at a new steady state within ∼30 s.42
) response usually stabilizes at a new steady state within ∼30 s.42

Cardiovascular compensatory and decompensatory responses to increasing severity of hypoxemia comparing young/healthy subjects to both elderly subjects and those with cardiovascular disease. HR = heart rate; CO = cardiac output; PVR = pulmonary vascular resistance; SVR = systemic vascular resistance. From Reference 43, with permission.
In moderate hypoxemia (PaO2 40–50 mm Hg, SaO2 75–80%), young adults continue to respond with increased heart rate, cardiac output, and increases in both pulmonary and systemic vascular resistance (ie, increased pulmonary arterial and systemic arterial blood pressure). In contrast, both aged adults and those with cardiovascular disease exhibit a less intense cardiovascular response. Consequently, metabolic acidosis may become apparent, and the risk of cardiovascular failure increases substantially.43
When hypoxemia is severe (PaO2 30–40 mm Hg, SaO2 50–75%), young healthy adults respond with substantially increased tachycardia and cardiac output, with acidemia becoming a prominent feature. Acidemia worsens even further in aged adults and those with limited cardiovascular compensatory reserves. Under these conditions, the risk of end-organ damage, acute cardiac injury, and cardiovascular collapse increases substantially.43 Profound hypoxemia (PaO2 < 30 mm Hg, SaO2 < 50%) occurring clinically portends precipitous cardiovascular collapse that typically results in loss of consciousness, bradycardia, and shock.43
Severe Hypoxemia and Cardiorespiratory Decompensation in COVID-19
During the early months of the pandemic, incidences of happy hypoxia coinciding with sudden, catastrophic hemodynamic collapse only amplified concerns regarding silent hypoxemia. That these concerns lead to mistaken comparisons with acute high-altitude hypoxic exposure obliges further examination of this issue, as both phenomena have a partial basis in physiology.
As described above, the normal response to an acute change in altitude and hypoxic hypoxia includes both hyperventilation and a hyperdynamic cardiac response. Here we refer to a rapid change as might occur in an unpressurized aircraft or even running the Pikes Peak marathon (ascent to ∼14,200 ft) compared to acclimatization of individuals at high altitudes over prolonged periods of time.
The acute cardiorespiratory response preserves oxygen delivery coincident with the degree of hypoxemia. The simultaneous increase in 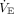 reduces PaCO2 and increases pH, both increasing alveolar oxygen based on the alveolar air equation, causing a leftward shift in the oxyhemoglobin disassociation (higher SaO2 for a given PaO2).14,22
reduces PaCO2 and increases pH, both increasing alveolar oxygen based on the alveolar air equation, causing a leftward shift in the oxyhemoglobin disassociation (higher SaO2 for a given PaO2).14,22
In normal subjects at high altitude with normal lung mechanics, hyperventilation often manifests itself as a reduced breathing frequency and large tidal volume (VT > 1.0 L). This response is far different than that of a patient with viral pneumonia and reduced respiratory system compliance (CRS). In fact, hypoxemia is tolerated well by most individuals, yet the combination of hypoxemia and cardiovascular collapse (signifying loss of compensation) results in ischemia and anoxic injury. However, when cardiac output is normal or elevated, hypoxia loses its ability to wreck the machinery.
In contrast, when hypoxemia occurs in older patients and those with cardiovascular disease, hemodynamic and pulmonary compensation is limited. This is the basis for observations that patients with COVID-19 appeared to be “happy hypoxics” just prior to catastrophic collapse.48,49 Thus, any preexisting disease that limits cardiac output hinders compensation. As underlying disease progresses and hypoxemia worsens, acidemia ensues, leading to cardiac failure and death.50,51
Sudden deterioration in both oxygen saturation and cardiovascular compensation may occur rapidly when hypoxemia primarily results from intrapulmonary shunt.52 The combination of increased intrapulmonary shunt and a fall in 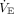 and cardiac output, coupled with an SaO2-PaO2 resting on the steep part of the oxygen hemoglobin disassociation curve, portends impending failure. A reduction in cardiac output worsens hypoxemia through a decrease in mixed venous oxygen. Acidosis causes a right shift in the oxyhemoglobin disassociation curve, thus defeating compensatory mechanisms. On the steep portion of the oxyhemoglobin disassociation curve, minor changes in PaO2 result in substantial changes in SaO2. These tenuous relationships may explain the signs of rapid deterioration seen in subjects with COVID-19 (Table 1).
and cardiac output, coupled with an SaO2-PaO2 resting on the steep part of the oxygen hemoglobin disassociation curve, portends impending failure. A reduction in cardiac output worsens hypoxemia through a decrease in mixed venous oxygen. Acidosis causes a right shift in the oxyhemoglobin disassociation curve, thus defeating compensatory mechanisms. On the steep portion of the oxyhemoglobin disassociation curve, minor changes in PaO2 result in substantial changes in SaO2. These tenuous relationships may explain the signs of rapid deterioration seen in subjects with COVID-19 (Table 1).
Physiologic Changes Associated With Loss of Cardiovascular Compensation During Hypoxemia
Respiratory Drive in Response to Hypercapnia and Acidosis
Absence of a PaO2 chemosensitive response until a threshold of ∼60 mm Hg is reached reflects the fact that PaCO2 and [H+] are the most potent ventilatory stimuli and thus are the most tightly controlled variables during ventilation.53 This is partly explained by the fact that CO2 (possessing a higher solubility at a similar molecular weight) is ∼20 times more diffusible across tissues than O2.54 Thus, alterations in metabolism/respiration are detected much more rapidly through CO2 chemosensory pathways.
The response to acidosis does not differ between respiratory and metabolic origins22 and is detected by peripheral and central chemoreceptors.55 During the initial response to severe metabolic acidosis, the peripheral chemoreceptors are more important. In addition, when PaCO2 increases and/or pH decreases, the carotid body receptors also become increasingly sensitive to hypoxemia.
In addition, during normal respiratory cycles, peripheral CO2 chemoreceptor output varies synchronously with small PaCO2 oscillations. They are more sensitive and respond faster than central receptors to sudden changes in PaCO2 (eg, during exercise).22,30 Peripheral and central CO2 chemoreceptors work in concert so that peripheral receptor stimulation amplifies the corresponding output of the central receptors.54
The relationship between PaCO2 and respiratory drive is signified by  response curves that are linear and steep (slope of 2 L/min per mm Hg) at a normal PaO2. This acuity in PaCO2 control is observed in both the early stages of sleep and during mild-to-moderate exercise when PaCO2, respectively, increases or decreases by only 1–3 mm Hg.56,57 The curves also become steeper in response to severe hypoxemia (PaO2 ∼40 mm Hg) or severe metabolic acidosis.22
response curves that are linear and steep (slope of 2 L/min per mm Hg) at a normal PaO2. This acuity in PaCO2 control is observed in both the early stages of sleep and during mild-to-moderate exercise when PaCO2, respectively, increases or decreases by only 1–3 mm Hg.56,57 The curves also become steeper in response to severe hypoxemia (PaO2 ∼40 mm Hg) or severe metabolic acidosis.22
Respiratory Drive Variability: Neurotransmitter and Genetic Considerations
The hypoxic ventilatory response varies considerably between individuals. Some react with substantial increases in breathing frequency and/or VT, whereas others exhibit little response. The accompanying cardiovascular response to hypoxemia shows similar interindividual variability.
Such variability is thought by some to reflect central neurotransmitter production and release (or accumulation) over time.58 During acute or chronic hypoxemia, the excitatory neurotransmitter glutamate increases ventilatory demand.59 As glutamate levels rise, so too does its conversion to gamma aminobutyric acid, a neurotransmitter that depresses ventilation. The biphasic ventilatory response to acute hypoxemia (described below) likely signifies the interplay of these neurotransmitters and perhaps reflects interindividual genetic differences in their expression.
Genetic variation in respiratory drive is suspected to account for interindividual differences found among diverse high-altitude populations around the world. Potentially over 1,000 genes might be involved in the adaptation to chronic hypoxemia.60 Suspected genetic differences may account for the ∼1–33% of various high-altitude populations who reportedly suffer from chronic mountain sickness (Monge disease), which breathlessness is a common symptom.60
In contrast, hypercapnia increases respiratory drive primarily through the excitatory effects of acetylcholine, the primary neurotransmitter governing basic rhythmicity.59 There is emerging evidence possibly linking both sudden infant death syndrome and congenital central hypoventilation syndrome to mutations in the “ret” proto-oncogene responsible for prenatal neuronal development of CO2 chemosensitive pathways in the brain.59
Respiratory Drive in ARDS
The majority of patients with COVID-19-associated acute respiratory failure has or eventually develops ARDS.61 Elevated respiratory drive in ARDS is multifactorial, and it is impossible to gauge the specific contributions of any one sensory input. Common characteristics associated with ARDS include rapid shallow breathing and vigorous inspiratory effort.62-64 As described below, these pathologic alterations increase respiratory drive and contribute to the sensation of dyspnea and breathlessness (as described below).
Lung inflammation also contributes to respiratory drive and altered breathing pattern. Both hydrostatic and altered permeability pulmonary edema, as well as endogenous substances (eg, histamine and prostaglandins), stimulate alveolar juxta-pulmonary capillary receptors (J receptors). Stimulation of these irritant J receptors is associated with falling lung compliance that induces rapid shallow breathing.35,65,66
Stimulation of slow-adapting alveolar mechanoreceptors induces the Hering-Breuer deflation reflex, causing tachypnea and increased inspiratory force. These mechanoreceptors typically respond to sudden pronounced lung deflation, wherein the response intensity is proportional to the severity of lung collapse.37 Although typically associated with pneumothorax, the deflationary reflex theoretically might enhance respiratory drive during an acute loss of functional residual capacity from congestive/compressive atelectasis (eg, fulminant ARDS).
Decreased CRS with rising 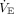 requirements in ARDS increases respiratory drive and work of breathing.67,68 As an example, when CRS is markedly reduced in ARDS, spontaneous rapid shallow breathing patterns are strongly associated with respiratory drive, peak inspiratory effort, and the magnitude of
requirements in ARDS increases respiratory drive and work of breathing.67,68 As an example, when CRS is markedly reduced in ARDS, spontaneous rapid shallow breathing patterns are strongly associated with respiratory drive, peak inspiratory effort, and the magnitude of 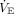 deficit (ie, the difference between what subjects can generate on their own during unassisted breathing vs what they can achieve during assisted mechanical ventilation).68 In this particular study, a large
deficit (ie, the difference between what subjects can generate on their own during unassisted breathing vs what they can achieve during assisted mechanical ventilation).68 In this particular study, a large  deficit (∼3.5–6.0 L/min) likely reflected the additional effects of acute hypercapnia. This exemplifies what others have described in ARDS: the disparity between neural demand and respiratory muscle capacity (under loaded conditions) in the context of increased metabolic demand (metabolic hyperbola).69
deficit (∼3.5–6.0 L/min) likely reflected the additional effects of acute hypercapnia. This exemplifies what others have described in ARDS: the disparity between neural demand and respiratory muscle capacity (under loaded conditions) in the context of increased metabolic demand (metabolic hyperbola).69
Respiratory Drive and the Theory of Minimal Work
Although rapid shallow breathing in ARDS may reflect input from afferent lung receptors, the pattern is consistent with the theory of minimal work proposed by Otis.70 The theory posits that the central respiratory pattern generator selects a VT and rate that minimizes the respiratory muscle power output needed to achieve sufficient 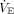 to maintain gas exchange homeostasis. When CRS is low, the most energy-efficient breathing pattern consists of a smaller VT to minimize the elastic work of breathing (and, therefore, dyspnea), compensated for by an elevated breathing frequency.
to maintain gas exchange homeostasis. When CRS is low, the most energy-efficient breathing pattern consists of a smaller VT to minimize the elastic work of breathing (and, therefore, dyspnea), compensated for by an elevated breathing frequency.
The minimal work theory is supported by laboratory research in healthy humans demonstrating that respiratory muscle fatigue and muscle failure occur when the combined inspiratory force generated by all inspiratory muscles during tidal ventilation exceeds 50–70% of their maximal force capacity.71 In ARDS, respiratory muscle weakness also is prevalent, as is increased elastic work of breathing and 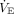 demand. Hence, rapid shallow breathing can be construed as an adaptive survival mechanism that likely minimizes the sensation of dyspnea.
demand. Hence, rapid shallow breathing can be construed as an adaptive survival mechanism that likely minimizes the sensation of dyspnea.
Breathing Perception and Dyspnea
It is generally accepted that respiratory drive and dyspnea are intertwined as they share the same afferent receptors and are processed by the same central neural pathways.72 Therefore, any mechanism that increases or reduces respiratory drive most likely has a similar impact on the perception of effort and the generation or amelioration of dyspnea.
Definitions and Nuanced Distinctions
Dyspnea is a general term describing difficulty or unpleasantness in the act of breathing. Similar to pain, dyspnea possesses qualitatively distinct features of varying intensity processed by the same brain structures73 so that both sensations likely produce similar degrees of suffering.
Dyspnea also is used in a specific manner to describe the perception that inspiratory effort is disproportionately greater than (out of balance with) the corresponding degree of simultaneous chest expansion.74 Likewise, breathlessness specifically denotes an awareness of excessive ventilatory drive or an “unpleasant urge to breathe.”74 This manifests either as an urge to breathe that cannot be met (eg, feeling winded) or situationally inappropriate (eg, elevated ventilation at rest). Although in its narrow usage dyspnea is associated with loaded breathing and breathlessness with chemosensory stimulation (hypoxic or hypercapnic), both sensations may be experienced simultaneously (eg, when severe metabolic acidosis, severe hypoxemia, and loaded breathing occur together in ARDS).75
Although confusing at times, dyspnea is most often used in its general sense as matter of convenience. The context in which the term is used often provides hints as to its intended specificity. For example, dyspnea is frequently substituted for breathlessness when the sensory effect of either hypoxemic or hypercapnic chemosensory stimulation is being described.
Other sources of dyspnea include J-receptor activation during pulmonary edema65 and mechanoreceptor stimulation during acute lung volume loss.37 Key to all these sensations is the sense of alarm generated by the awareness of an abnormal effort to breathe (the awareness of respiratory drive). Finally, the limbic/paralimbic system may cause breathlessness. This may occur indirectly with anxiety-induced hypocapnia that stimulates the amygdala or directly through hypercapnic-induced stimulation of the entire limbic/paralimbic system.76
Beyond these archetypical descriptors exists qualitatively distinct sensations commonly associated with specific cardiopulmonary diseases such as chest tightness (asthma), gasping (interstitial pulmonary fibrosis), burning (bronchitis), and suffocation (congestive heart failure). This varied and nuanced language often coincides with other descriptors that may change over the course of cardiopulmonary or neurologic disease as the mechanics of breathing and chemosensory input change.77
The Theory of Length-Tension Inappropriateness
Dyspnea as mechanical difficulty in the act of breathing was conceived initially as an error-correcting proprioceptive mechanism, one that is generated by muscle spindle fibers during resistive, elastic, or threshold loading (ie, length-tension inappropriateness).75 Abrupt loading increases muscle tension disproportionately greater than the corresponding, instantaneous degree of muscle shortening. This creates misalignment between parallel force-generating and stretch-stabilizing muscle fibers, which is sensed by afferently innervated connective tissue residing between these 2 fibers called muscle spindles. Muscle spindle activation stimulates a reflexive correction occurring at the medullary-pontine level. During the same or subsequent breaths, increasing inspiratory effort (muscle tension) corrects the error to achieve the targeted VT (ie, chest displacement via muscle shortening), ensuring stable ventilation.
Because dyspnea denotes conscious awareness, critical thresholds of afferent stimuli associated with respiratory drive (ie, the summation of inputs from length-tension proprioceptors, CO2 and O2 chemoreceptors, and mechanical and irritant receptors) project up to higher brain centers (ie, thalamus, limbic/paralimbic, and sensory/motor cortices). This is achieved by stimulation of a diffusive web of regulatory neurons within the reticular formation (ie, medulla, pons, and upper cervical spinal cord) that mediates reflexive and other nonconscious vital functions (Fig. 1).
Dyspnea as Neuromechanical Dissociation
Although the theory of length-tension inappropriateness was paradigmatic for studying dyspnea in the 1960s, its vagueness regarding the governing mechanism once dyspnea is perceived reflected the limited knowledge at that time. Approximately 40 years later, a new iteration of the theory described dyspnea as neuromechanical dissociation or efferent-reafferent–dissociation signaling,78-80 whereby dyspnea constitutes an unexpected event.81
Disturbing respiratory sensations, as well as the response to them, are governed primarily by interactions between the somatosensory and motor cortices. This occurs through the mechanism of corollary discharge that describes the cross-communication between these 2 structures. Once aroused, both the sensory and motor cortices take executive control over respiratory drive (eg, “I feel like I have to concentrate on my breathing”). Efferent impulses from the motor cortex are sent in parallel to both the medullary centers and the somatosensory cortex. The somatosensory cortex in turn interprets (compares) the strength of efferent impulses to the strength of integrated afferent impulses it receives simultaneously. Thus, the conception of dyspnea has evolved from length-tension inappropriateness to efferent-reafferent–dissociation signaling (Fig. 1).
Hypoxemic-Induced Dyspnea
Healthy subjects exposed to hypoxemia appear unable to detect altered breathing sensations between SpO2 80% versus 90%,82 and hikers ascending to ∼14,200 ft (eg, Pikes Peak, Colorado, estimated PaO2 ∼40 mm Hg) may or may not experience dyspnea.83
This marked variability in hypoxemia’s dyspnogenic potency was illustrated using experimental data on hypoxemia-induced dyspnea at rest. At the cusp of severe hypoxemia (ie, PaO2 40 mm Hg, PaCO2 40 mm Hg), an estimated 30% of individuals would not experience significant dyspnea.8 Even during severe hypoxemia (SaO2 < 70%), individual responses have ranged from profound dyspnea and panic to calmness and a sense of well-being.43 Others reported that if subjects were allowed to set their own 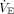 breathlessness was virtually undetectable at PaO2 40–45 mm Hg.84 And even when instructed to constrain their
breathlessness was virtually undetectable at PaO2 40–45 mm Hg.84 And even when instructed to constrain their 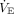 to resting levels, severe air hunger was not experienced by 50% of subjects.
to resting levels, severe air hunger was not experienced by 50% of subjects.
Hypoxic Ventilatory Decline as Happy Hypoxia
The absence of distress during hypoxemia is partly accounted for by hypoxic ventilatory decline. Just as there are mechanisms that stimulate the ventilatory response to hypoxemia, there also exist inhibitory mechanisms. Ventilatory response to hypoxemia also changes over time and may decline within as little as 15–20 min, becoming increasingly periodic despite worsening or persistent hypoxemia (Fig. 4).43

Representation of hypoxic ventilatory response signified by a sudden, initial rise and minute ventilation (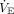 ) and subsequent cessation or reduction to a plateau somewhere above the initial baseline (ie, hypoxic ventilatory decline). The secondary rise over a period of weeks represents the process of acclimatization to living at higher altitudes (eg, 14,000 ft above sea level). Adapted from Reference 43.
) and subsequent cessation or reduction to a plateau somewhere above the initial baseline (ie, hypoxic ventilatory decline). The secondary rise over a period of weeks represents the process of acclimatization to living at higher altitudes (eg, 14,000 ft above sea level). Adapted from Reference 43.
A clinically relevant example of hypoxic ventilatory decline was reported by Easton et al58 describing a biphasic ventilatory response to sustained hypoxemia over ∼30 min. Moderate hypoxemia (SaO2 80%) under isocapnic conditions caused an immediate ∼60% rise in  (from 8 to ∼13 L/min). However, within ∼5 min
(from 8 to ∼13 L/min). However, within ∼5 min 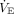 declined to a plateau ∼20% above baseline. This suggests that a relatively rapid inhibitory effect upon respiratory drive occurs and functions independently of PaCO2. This likely reflects the homeostatic interplay between excitatory-depressive neurotransmitters as the sudden, large release of glutamate also increases gamma aminobutyric acid levels, thus creating a biochemical brake that establishes a new, albeit higher equilibrium.
declined to a plateau ∼20% above baseline. This suggests that a relatively rapid inhibitory effect upon respiratory drive occurs and functions independently of PaCO2. This likely reflects the homeostatic interplay between excitatory-depressive neurotransmitters as the sudden, large release of glutamate also increases gamma aminobutyric acid levels, thus creating a biochemical brake that establishes a new, albeit higher equilibrium.
In passing, the actual sense of profound well-being is distinct from the mere absence of distress. The former initially was described in 1875 by Gaston Tissandier, the lone survivor of a tragic high-altitude ballooning experiment. At ∼23,000 ft, he experienced an overall sense of “oppression,” quickly relieved by periodically breathing from a bag containing 60% O2. But at ∼25,000 ft (estimated PaO2 < 20 mm Hg), he reported a “numbness of experience” with the “mind weakened little by little” in which he experienced “rising, inner joy” and “indifference” to the danger of which he was cognizant.74 Similar reports regarding trekking accidents on Mount Everest (∼29,000 ft) and similar high-altitude peaks have been attributed to hypoxia-induced “poor judgment.”85
Experiences of calm and well-being have been reproduced in hypobaric simulation studies when hypoxemia is accompanied by hyperventilation.44 In one such simulation of 30,000 ft resulting in PaO2 22–28 mm Hg and PaCO2 16–31 mm Hg, the vast majority of subjects (89%) was alert and cooperative, with no signs of respiratory distress. However, if euphoria was experienced, it apparently was not salient enough to merit mentioning by the investigators.86
Hypercapnia and Breathlessness
It bears repeating that unless PaO2 is ∼45 mm Hg hypoxemia alone often does not induce dyspnea, particularly when PaCO2 is < 40 mm Hg.8,40 A study examining how acute hypercapnia generates breathlessness in normal subjects found that by increasing PETCO2 from 39–43 mm Hg only a slight sensation of breathlessness was experienced.87 In contrast, breathlessness intensified rapidly as PETCO2 rose to 45–48 mm Hg, becoming intolerable at ∼50 mm Hg. In another study, the threshold of severe breathlessness occurred at PETCO2 ∼10 mm Hg above baseline.88 Given that PETCO2 is normally ≤ 5 mm Hg < PaCO2, it suggests that mild breathlessness is induced at a maximum PaCO2 ∼48 mm Hg, increasing in intensity between 50–53 mm Hg and becoming intolerable at a maximum PaCO2 ∼55 mm Hg.
Dyspnea in the Laboratory Setting: Interplay Between Hypoxemia, Hypercapnia, and Hypocapnia
The stimulatory weakness of hypoxemia and its modulation by the corresponding PaCO2 have been elegantly illustrated in other laboratory studies of dyspnea. In one study, an acute drop in PaO2 from 96 to 47 mm Hg barely registered as breathlessness on intensity rating scales when PaCO2 was 35 ± 5 mm Hg.47 Another found a 10 mm Hg decrement in PaCO2 (∼30 mm Hg) essentially abolished hypoxemic-induced breathlessness.89 Thus, at least modest-to-moderate degrees of hypocapnia suppress dyspnea as part of the hypoxic ventilatory response.
The intensity of breathlessness also has been compared using different inspired gas mixtures (ie, hyperoxic and hypoxic mixtures combined with hypercarbic and hypocarbic mixtures).74 Breathlessness was most intense when breathing a hypercarbic-hypoxic gas mixture and was reduced slightly when breathing a hypercarbic-hyperoxic gas mixture. And in relevance to silent hypoxemia, only modest breathlessness was observed while breathing a hypoxic-hypocarbic gas mixture.
Hypoxemia and Neuromechanical Dissociation
When considering hypoxemia as a source of dyspnea, it is noteworthy that a precipitous decline in SaO2 to 80% increases peak inspiratory muscle pressure by ∼8 cm H2O (manifested by increased VT rather than frequency).89 This represents a very small fraction of normal inspiratory muscle pressure reserve (≥ 120 cm H2O).71,90,91
Perception of dyspnea during mechanical loading is best expressed as the ratio of pressure generated during tidal breathing relative to inspiratory muscle pressure reserve (PI/PI-max), with the intensity of dyspnea increasing linearly with the fractional increase in effort.92-94 The perception of severe effort occurs when PI/PI-max is ≥ 50%.95 Applying these proprioceptive findings to the early phase of COVID-19 (ie, when functional residual capacity, CRS, and muscle strength are relatively well preserved) suggests the likelihood of dyspnea associated with hypoxemia-induced ventilatory demand is likely minor (see COVID-19 Type L ARDS below).
Silent Hypoxemia and COVID-19: Overview and Case Reports
As described in the introduction, initial reports from China mentioned silent hypoxemia only in passing. The largest study to do so (∼1,100 cases) reported shortness of breath in 205 subjects (∼19%), the majority of whom (68%) described it as mild. 96 Curiously, some interpreted these findings as reflecting the prevalence of silent hypoxemia.6,97 Yet the veracity of these data is indeterminant. It was largely based upon evaluations done at hospital admission. Therefore, it likely missed hypoxemic subjects in whom dyspnea subsequently developed as pneumonia worsened. More importantly, the presence or absence of dyspnea was not correlated to concurrent assessments of oxygenation, ventilation, breathing pattern, or radiographic presentation.
A clearer perspective was provided by 6 studies published in 2020 (N ∼1,700 subjects). These reports recorded baseline symptoms and found dyspnea occurred frequently, ranging between 35–91% with an average incidence of 61%.98-103 In a prospective study of 30 hospitalized subjects with mild COVID-19 (ie, not requiring ICU admission), 73% complained of dyspnea, all of whom had normal or heightened alveolar ventilation.104
We found 7 case reports/case series describing apparent silent hypoxemia in a total of 12 subjects. Arterial blood gas (ABG) data accompanied SpO2 measurements in 8 subjects (Table 2). In 2 subjects, SpO2 was ≥ 85% (mild hypoxemia),5,13 and in 7 SpO2 was ≤ 75% (severe hypoxemia).7,14,105,106 Among 3 subjects whose SpO2 ranged from 60–66%, their corresponding PaO2 was substantially > predicted (51–56 mm Hg).105 Among 8 subjects with ABG data, PaCO2 was reported in only 3, each of whom exhibited mild-to-moderate hyperventilation. Interestingly, in 2 other subjects with supposed silent hypoxemia, one was actually experiencing mild dyspnea and was tachypneic,12 whereas the other was never directly questioned about experiencing any breathing discomfort.5
Case Reports, Brief Communication, and Observations From Studies Not Specifically Examining the Phenomenon of Silent Hypoxemia in COVID-19
Silent Hypoxemia and COVID-19: Case-Controlled Studies
A retrospective case-control study (N = 213) extracted data from electronic information systems at 2 Italian hospitals during the first wave of the pandemic.16 Given the enormity of the crisis, the ability of clinicians to assess the quality and intensity of dyspnea understandably was limited, as was establishing baseline hypoxemia prior to initiating O2 therapy. Data were absent in ∼50% of subjects. Approximately 32% of the remaining subjects were judged to have silent hypoxemia. Those without dyspnea were significantly less hypoxemic than dyspneic subjects (PaO2/FIO2 225 ± 68 mm Hg vs 192 ± 78 mm Hg, P = .002), and both cohorts exhibited mild hyperventilation (PaCO2 34 ± 7 mm Hg vs 34 ± 6 mm Hg, respectively, P = .47). When analyzed according to chest radiographic abnormalities, non–dyspneic subjects represented the majority of those (∼55%) with normal chest radiographs compared to ∼25–35% with increasingly severe lung opacities.
Taken together, subjects with apparent silent hypoxemia had less severe lung injury and were observed either prior to developing ARDS or never developed the syndrome. The primary difficulty in interpreting this study is the absence of oxygenation differences between non–dyspneic and dyspneic cohorts in terms of PaO2 (66 ± 23 mm Hg vs 70 ± 30 mm Hg, respectively, P = .24). This suggests that many non–dyspneic subjects either never reached the hypoxemic threshold for dyspnea or dyspnea was ameliorated by compensatory hypocapnia.
In another retrospective case-control study of 82 hypoxemic subjects, severity of dyspnea (assessed with the Borg CR10 scale) was compared between subjects with COVID-19 and subjects with chronic cardiopulmonary disease and/or community-acquired pneumonia.107 Subjects with COVID-19 presented with minimal dyspnea and median (interquartile range [IQR]) Borg score 1 (∼0.5–2), translating into sensations described as just noticeable or weak. By contrast, subjects with chronic pulmonary disease tended toward pronounced dyspnea. Subjects with COVID-19 had a median PaO2 64 (IQR 61–66) mm Hg and median PaCO2 32 (IQR 31–36) mm Hg, respectively, with 71% having PaCO2 < 35 mm Hg. Thus again, apparent silent hypoxemia in subjects with COVID-19 could be explained by both the lack of sufficient hypoxemic stimulus and/or compensatory hyperventilation.
A prospective case-controlled study15 represents the best evidence available to date because of 2 crucial design features: Enrollment required a baseline SpO2 < 80%, and variability in assessing dyspnea was minimized by mandating use of the American Thoracic Society definition (Table 3).108 In brief, only 5% of hypoxemic subjects with a median room air SpO2 76% (IQR 60–79) presented with apparent silent hypoxemia, with 91% of these cases also presenting with tachypnea. These subjects had similar degrees of hypocapnia, with median PaCO2 33 (IQR 30–37) mm Hg. These findings are consistent with those reported in healthy, severely hypoxemic research subjects experiencing minimal dyspnea when allowed to set their own PaCO2.47
Data from Garcia-Grimshaw et al Prospective Case-Control Study Comparing Dyspneic Versus Non–Dyspneic Subjects With COVID-19 Presenting With Hypoxemia
Despite similar degrees of lung injury severity (based on chest computed tomography findings) and a similar PaO2/FIO2 on the same FIO2, non–dyspneic subjects differed from dyspneic subjects in 3 respects: (1) They presented to the hospital 2 d earlier from symptom onset; (2) had a higher median room air SpO2, 76% (IQR 60–79) versus 70% (IQR 57–76), respectively, (P = .02); and (3) had a lower median breathing frequency, 22 (IQR 20–26) versus 30 (IQR 26–36), respectively, (P < .001).15 Finally, neither age nor co-diagnosis of diabetes differed between cohorts, which might have explained the absence of dyspnea (as described below).
Finally, a smaller prospective case-control study examined subjects with non–severe COVID-19 for dyspnea, neurologic symptoms, and respiratory-related variables.109 As assessed by structured interviews coupled with Borg scale evaluation, the incidence of dyspnea was 32%. Neither the presence of neurologic symptoms (eg, loss of smell or taste) nor respiratory frequency and ABG findings differed between dyspneic and eupneic subjects.
Non–Neurogenic Explanations for Silent Hypoxemia in COVID-19
“Whenever possible, substitute constructions out of known entities for inferences to unknown entities.”
Bertrand Russell110
Several opinion pieces and reviews offered various perspectives on why some patients with COVID-19 appear to have silent hypoxemia.4,8,14,43,44,111 To date, however, the only valid evidenced-based explanation for silent hypoxemia either in the absence of severe hypoxemia or when severe hypoxemia occurs with elevated PaCO2 is that hypoxemia is a relatively weak stimulus both for respiratory drive and dyspnea.22,43,112,113
Confusion regarding apparent COVID-19-associated silent hypoxemia sometimes has involved puzzling, unorthodox definitions of hypoxemia. Some reports105,106 mistakenly defined the hypoxemic threshold as PaO2 < 80 mm Hg (vs the experimentally established threshold of ≤ 60 mm Hg), thus attributing silent hypoxemia when it was not present.
Regarding ARDS (which the majority of hypoxemic patients with COVID-19 develops),114 there is a tendency to interpret the coincidence of hypoxemia with dyspnea as representing a cause-effect relationship. However, severe neuromechanical dissociation is the most likely source of dyspnea in ARDS. This occurs alongside less salient but key factors such as stimulation of afferent receptors in the pulmonary tissue as well as suprapontine inputs representing the psychological and emotional trauma associated with respiratory distress and critical illness.115,116
We suspect clinicians tend to associate low SpO2 with dyspnea because of 3 factors. First, the almost reflexive focus on oxygenation when evaluating patients with respiratory disease. Second, the divergence between what physiologists versus clinicians consider to be severe hypoxemia (eg, SaO2 ∼75% vs ∼85%, respectively). And third, the pervasive presence of SpO2 visual stimuli in the clinical setting constantly reinforces the focus upon oxygenation. These factors likely divert attention away from less salient but more potent dyspnogenic stimuli. In consequence, clinicians tend to overestimate the dyspnogenic impact of hypoxemia. Given the stressful, chaotic circumstances during the pandemic, the expectation that hypoxemia and dyspnea coexist as a cause-effect relationship made its absence all the more conspicuous and perplexing. Thus, it was perceived as another manifestation of an unfamiliar and deeply frightening contagion.
Impact of COVID-19 Type L ARDS on Dyspnea
A cogent explanation for apparent silent hypoxemia during COVID-19 involved the underlying pathophysiology during the early stages.4,43 Gattinoni and colleagues described this as Type L (atypical ARDS) whereby coronavirus infection of the pulmonary vascular endothelium abolished compensatory hypoxemic pulmonary vasoconstriction. This caused profound ventilation-perfusion mismatching and severe hypoxemia, despite near-normal CRS, functional residual capacity, and modest degrees of lung inflammation.117 Type L ARDS also was hypothesized as causing self-inflicted lung injury. This presupposes both intact respiratory muscle strength and normal PI/PI-MAX proprioceptive relationships described earlier. Therefore, the ability to suppress or ameliorate dyspnea under Type L conditions only requires the ability to achieve modest hypocapnia at negligible increases in effort.
We calculated the corrected 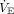 118 from a large COVID-19 study (N = 267) that published
118 from a large COVID-19 study (N = 267) that published  and corresponding PaCO2 data.119 Study subjects in the 25th and 50th quartiles had an initial corrected
and corresponding PaCO2 data.119 Study subjects in the 25th and 50th quartiles had an initial corrected 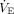 (ie, that needed to achieve PaCO2 40 mm Hg)118 that was ≤ 7.7 L/min and ≤ 10.3 L/min, respectively. Modestly higher
(ie, that needed to achieve PaCO2 40 mm Hg)118 that was ≤ 7.7 L/min and ≤ 10.3 L/min, respectively. Modestly higher 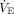 levels needed to achieve mild hypocapnia in a large number of these subjects would appear highly plausible under Type L conditions. Likewise, the corresponding respiratory muscle power output and central drive needed to achieve suppressive PaCO2 (ie, 35 ± 5 mm Hg)47 would be negligible.
levels needed to achieve mild hypocapnia in a large number of these subjects would appear highly plausible under Type L conditions. Likewise, the corresponding respiratory muscle power output and central drive needed to achieve suppressive PaCO2 (ie, 35 ± 5 mm Hg)47 would be negligible.
Natural Variations in Control of Breathing, Comorbidities, and Altered Mental Status
Others have advanced equally compelling explanations for apparent silent hypoxemia not requiring coronavirus infection of the peripheral and/or central and nervous systems.8,43 First, there exists a 10-fold difference in respiratory drive in how individuals respond to hypoxemia and hypercapnia, supported by physiologic research into natural variations in respiratory drive among high-altitude populations. Second, both older individuals and those with diabetes have blunted ventilatory response to chemoreceptor stimulation.49,120 Studies of older subjects (64–73 y old) found respiratory drive responses to both hypoxemia and hypercapnia are reduced by 40–50% compared to young adults (22–30 y old).48,49
This information is particularly important in assessing silent hypoxemia during the first wave of COVID-19, when hospitalized subjects with COVID-19 largely were older and/or had diabetes as a comorbidity. In one study, 44% of subjects was ≥ 65 y old, and 37% had diabetes,98 whereas in other studies 23–54% of subjects was ≥ 70 y old, and 17–28% had diabetes.121,122 Hence, a sizable percentage of hospitalized subjects with COVID-19 likely had blunted hypoxemic ventilatory response at baseline. The tendency toward blunted hypoxic drive during the first wave of COVID-19 far exceeded the actual incidence of apparent silent hypoxemia (5%) reported in the largest, best-controlled prospective study specifically focused on this phenomenon.15
Finally, acutely ill hypoxemic patients often have altered mental status that can mask symptoms.8 This makes the veracity of diagnosing silent hypoxemia all the more problematic, particularly so when trying to evaluate and triage patients in the chaotic environment that was the early months of the pandemic. In such circumstances, the careful, time-consuming evaluation required to accurately assess dyspnea was at best impractical if not impossible.115
Accuracy of Pulse Oximetry and Apparent Silent Hypoxemia
Within the construct of diagnosing hypoxemia, a return to the principles that govern the relationship of PaO2 and SaO2 as well as factors impacting the accuracy of pulse oximetry is in order. Pulse oximeters are ubiquitous in all health care facilities and perhaps deceptively simple. Our intent here is not to cover all the factors that impact oximetry accuracy but rather to highlight those seen with COVID-19 that might suggest a lower SpO2 than is actually present.123
The PaO2–SaO2 relationship is described by the sigmoid shape of the oxyhemoglobin disassociation curve.124 Under normal physiology, SaO2 90% is typically associated with PaO2 60 mm Hg. Over decades, the mnemonic “30–60, 60–90, 40–75” has aided clinicians as a rule of thumb for remembering the PaO2-SaO2 relationship. With alterations in physiology, associated changes in temperature, PaCO2, 2–3 diphosphoglycerate, and pH alter the normal PaO2-SaO2 relationship.
COVID-19 results in ARDS and profound hypoxemia but also often results in viral sepsis. With viral sepsis, both body temperature changes and hypotension (impacting signal quality) conspire to alter bedside oximeter accuracy. With respect to oxygenation status, a change in body temperature from 37°C to 40°C at a constant pH and PaCO2 will cause SaO2 to fall from 91% to 86%, a decrease of 5% for the same PaO2.14 At a lower PaO2, on the steeper portion of the oxyhemoglobin disassociation curve, the magnitude of change is greater. Clinically, this means a lower measured SpO2 displayed by the oximeter for a PaO2 value that does not meet the definition of hypoxemia or the suggested severity of hypoxemia. Clinically this is important in the discussion of silent hypoxemia, as carotid body chemoreceptors are sensitive only to PaO2, not SpO2.14
Pulse oximetry accuracy has been the focus of many publications and a major driver in this competitive market. However, the accuracy of pulse oximetry in critically ill mechanically ventilated patients is only ± 4%.125,126 These inaccuracies may be further compounded by the presence of hypotension and a consequent poor signal quality.127
Sjoding et al128 brought renewed attention to a well-known issue that oximetry accuracy is negatively impacted by skin pigment. Both the initial calibration of oximeters and the principle of operation (light through a tissue bed to a detector) account for greater inaccuracy. Jubran and Tobin129 described this effect back in 1990, to far less fanfare but important clinical impact. They found that SpO2 95% was required to assure PaO2 > 60 mm Hg in subjects with dark pigmentation. In one subject, SpO2 90% was associated with PaO2 49 mm Hg. Bickler and colleagues130,131 also detailed the impact of skin pigment on accuracy of oximeters across a spectrum of SpO2 values. Their studies predicted the findings by Sjoding et al nearly 20 years earlier, yet explanations for these results remain elusive.132
Neurologic Injury During COVID-19 as a Potential Source of Altered Perception of Dyspnea
“First Rule of Scientific Reasoning: We are to admit no more causes of natural things than such as are both true and sufficient to explain their appearance.”
Sir Isaac Newton133
Several investigators have questioned whether respiratory drive is altered by SARS-CoV-2 central nervous system infection, thus presenting a potentially valid alternative explanation for silent hypoxemia.18,19,21,134 All neurological infection hypotheses are based upon coronaviruses’ general ability to invade and live inside neural tissue (neurotropism).135 SARS-CoV-2 has been observed to infect the brains of both animals and patients, with infection of the medullary cardiorespiratory centers found to be particularly severe in one study.134
Alternatively, Tobin and colleagues14 suggested SARS-CoV-2 may infect the carotid bodies via endothelial angiotensin-converting enzyme 2 (ACE-2) receptors, thus blunting the ability of peripheral chemoreceptors to detect hypoxemia. Others have speculated that potential blunting of afferent stimuli might occur centrally in medullary structures receiving afferent signals from the carotid bodies (ie, via the nucleus tractus solitarius) (Fig. 1).18,21 In contrast, Gattinoni and colleagues136 proposed that SARS-CoV-2 infection of the carotid bodies might actually magnify hypoxic chemosensitivity, thus leading to patient self-inflicted lung injury.
Because ACE-2 receptors also are expressed in nasal mucosa, SARS-CoV-2 may enter the brain by infecting the olfactory bulb.134 This route of infection was demonstrated with transnasal inoculation in animal models using various coronaviruses.137,138 That anosmia and dysgeusia (loss of smell and taste) are often associated with COVID-19139 provides circumstantial support for neuroinvasive hypotheses. However, as others109 have observed, neither anosmia nor dysgeusia distinguishes subjects with COVID-19 based upon the presence or absence of dyspnea.
Mechanistically speaking, the most cogent explanation for silent hypoxemia among these hypotheses involves SARS-CoV-2 infection of the limbic system that governs emotions, particularly the amygdala, which plays a prominent role in generating fear and anxiety.140 Ultimately, the validity of this hypothesis is predicated upon uncovering evidence that SARS-CoV-2 infection of the amygdala (or other structures in the limbic system) actually exerts a depressive effect on generating stressful emotions. To our knowledge no such direct evidence exists.
Most pertinent is that histopathological evidence supporting a linkage between SARS-CoV-2 central nervous infection and silent hypoxemia is circumstantial at best and not particularly convincing. A postmortem histopathologic study of 20 subjects with COVID-19 tracing the route of brain infection found only a minority of subjects had definitive findings attributable to COVID-19.141 Only 20% of subjects had SARS-CoV-2 RNA detected in at least one area of the brain, with only the olfactory bulb testing positive in more than one subject. In contrast, SARS-CoV-2 RNA was found in the amygdala or the medulla in a single subject (5% incidence). Another histopathologic study of 20 subjects with COVID-19 focused exclusively on infection of both the olfactory bulb and the amygdala found substantially greater viral transcriptional changes in the olfactory bulb versus the amygdala.142
Although neurological infection hypotheses of silent hypoxemia are intriguing, and cannot be summarily dismissed, they are merely speculative and stand in stark contrast to a preponderance of established scientific evidence concluding that dyspnea is largely absent unless hypoxemia is severe.143,144
Summary
Silent hypoxemia is a well-described phenomenon that predates COVID-19 and can be explained by known human physiologic responses to hypoxemia as well as the nature of dyspnea. Whether this phenomenon is more common in COVID-19 has yet to be determined. But based upon the evidence accrued thus far, it appears unlikely. However, the reason it is being observed mostly can be explained.
The most important findings of this review of silent hypoxemia are that respiratory drive is not depressed in COVID-19, and the absence of dyspnea is largely explained by compensatory hyperventilation. Quite often the degree of hypoxemia documented in reports was insufficient to provoke dyspnea. Among subjects in well-designed prospective studies of silent hypoxemia, the actual incidence was low. In addition, current evidence supporting coronavirus infection of the central nervous system as a source of silent hypoxemia is indirect and circumstantial. This is in stark contrast to decades of physiologic research on hypoxemia, respiratory drive, and dyspnea.
Nonetheless, the term happy hypoxemia should be retired from the lexicon, as despite patients with seemingly low SpO2 and no overt signs of respiratory distress they are unlikely to have come to the hospital with viral pneumonia and remain happy. Understanding the underlying physiologic principles that govern these relationships is critical for adequate assessment of patients with hypoxemia and for critical care practice in general.
Footnotes
- Correspondence: Richard Kallet MSc RRT FAARC, 2070 Fell Street #1, San Francisco, CA. 94117-1878. E-mail: richkallet{at}gmail.com
Mr Kallet has received honoraria from ContinuED. Mr Branson discloses relationships with Engineered Medical Systems, Pfizer, Mallinckrodt, Ventec Life Systems, and Vyaire. Mr Branson is Editor in Chief of Respiratory Care. Dr Lipnick has disclosed no conflicts of interest.
- Copyright © 2022 by Daedalus Enterprises
REFERENCES
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.
- 100.
- 101.
- 102.
- 103.↵
- 104.↵
- 105.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 113.↵
- 114.↵
- 115.↵
- 116.↵
- 117.↵
- 118.↵
- 119.↵
- 120.↵
- 121.↵
- 122.↵
- 123.↵
- 124.↵
- 125.↵
- 126.↵
- 127.↵
- 128.↵
- 129.↵
- 130.↵
- 131.↵
- 132.↵
- 133.↵
- 134.↵
- 135.↵
- 136.↵
- 137.↵
- 138.↵
- 139.↵
- 140.↵
- 141.↵
- 142.↵
- 143.↵
- 144.↵





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Introduction
- Control of Breathing and Hypoxemic Cardiovascular Compensation
- Breathing Perception and Dyspnea
- Silent Hypoxemia and COVID-19: Overview and Case Reports
- Silent Hypoxemia and COVID-19: Case-Controlled Studies
- Non–Neurogenic Explanations for Silent Hypoxemia in COVID-19
- Neurologic Injury During COVID-19 as a Potential Source of Altered Perception of Dyspnea
- Summary
- Footnotes
- REFERENCES
- Figures & Data
- Info & Metrics
- References