

Abstract
This review describes the current understanding of the lungs' response to deforming stress under conditions of both normal physiology and acute lung injury. Several limiting assumptions are needed to infer lung parenchymal stress and strain from airway pressure, volume, and flow data from mechanically ventilated patients with injured lungs. These assumptions include the effects of the chest wall on lung-surface pressure, its topographical distribution, and the effects of non-uniform tissue properties on local parenchymal stresses. In addition, there is a spectrum of biophysical lung injury mechanisms that involves normal as well as tangential alveolar wall stresses. To these are added important secondary effects on pulmonary vascular resistance and right heart function. Understanding both the assumptions of lung mechanics and the scope of injury mechanisms operating during ARDS is necessary to interpret the results of clinical trials that inform prevailing ventilator-management guidelines. The implications issuing from these 3 topics inform a safer approach to setting and adjusting the ventilator to minimize the risk of ventilator-induced lung injury. This is enumerated in a 5-step approach that can be used to guide ventilator management of unstable patients with severe lung injury.
Introduction
The objective of this review is to put forth an approach to safer mechanical ventilation based on an understanding of factors that influence lung parenchymal stress and strain in disease. To this end, the limiting assumptions needed to infer lung parenchymal stress and strain from airway pressure, volume, and flow data of mechanically ventilated patients will be detailed, and the spectrum of biophysical lung injury mechanisms will be discussed. This knowledge will not only guide the interpretation of clinical trials, which to date have informed prevailing ventilator-management guidelines, but will also serve as the rationale for a 5-step outline for how clinicians may approach the management of unstable patients with severely injured lungs.
Stress and Strain in the Respiratory System
All modes of ventilator assistance share a common goal, namely, to support gas exchange while keeping lung parenchymal stress within tolerance limits. Stress defines a force per unit area, and as such has the same unit of measure as pressure. The pressure that most closely correlates with lung parenchymal stress is the elastic recoil pressure of the lung, which in a uniformly expanded lung is equal to the pressure difference between the alveoli and the lung surface (ie, the visceral pleura).1
While all prevailing ventilator-management guidelines seek to minimize parenchymal stress, lung stress is typically inferred because it is difficult to measure in situ for 3 reasons. First, the lung is non-uniformly expanded in the intact thorax, resulting in non-uniform pleural pressure and thus non-uniform parenchymal stress distributions.2 While esophageal manometry may succeed in identifying patients with very high regional pleural pressures, it would be misleading to extrapolate lung stress remote from the measurement site.3
Second, in injured lungs a significant number of alveoli are not in communication with the central airways, either because they are filled with fluid or occluded by liquid bridges and mucus at the level of conducting airways and alveolar ducts. In that instance, alveolar pressure, and thus local stress of closed units, cannot be inferred from airway occlusion pressure measurements.
Third, in a diseased lung there are stress concentrations between regions with heterogeneous mechanical properties. This network property was first described by Mead et al4 under the heading of parenchymal interdependence, and it reflects the tissue distortion between structures that resist deformation (eg, a flooded or collapsed alveolus) and their more pliable neighbors (neighboring alveolar walls) (Fig. 1).

Representation of shear stress developing within patent alveoli sharing common walls with a collapsed or non-expanding alveolus.
While parenchymal stress is an important determinant of injury risk, it is by no means the only one. Stress amplitude and its rate of change also shape the response of the lung to deforming stress.5 The importance of clinical ventilation parameters such as plateau pressure (Pplat), driving pressure (Pplat − PEEP), tidal volume (VT), inspiratory flow, and breathing frequency can only be understood in that context (Fig. 2). Driving pressure is analogous to stress amplitude, ie, the change in stress (or pressure) applied to a structure relative to its pre-stressed baseline state. A post hoc analysis of data from several large clinical trials suggested that driving pressure was the one variable that best stratified the biophysical lung injury risk of subjects with ARDS.6

Common clinical measurements and their biophysical correlates. VT = tidal volume, Pplat = plateau pressure.
For the purpose of that analysis, driving pressure was defined as the difference between extrinsic PEEP as set by the provider (assuming absence of inadvertent PEEP) and the end-inspiratory airway occlusion (ie, Pplat). This definition first discounts dynamic (loss) components of stress attributable to the rate of deformation, and second includes elastic pressures (stored energy) attributable to the elastic deformation of the passive chest wall. Therefore, it is best to refer to it as static respiratory system driving pressure or ΔP. These limiting assumptions should not distract from the importance of the observation by Amato et al6 insofar as it suggests that between-patient variability in chest-wall mechanics and dynamic energy dissipation may play a lesser role as injury risk factors.
While there is sound theoretical and some experimental evidence that the rate and frequency of lung deformation (ie, flow and frequency settings) modify injury risk, there are few if any compelling human data on that subject.7 The rationale to limit ventilator frequency settings to ≤ 30–35 cycles/min is largely based on diminishing rate effects on alveolar ventilation, while the practice to impose decelerating as opposed to square wave flow profiles is largely guided by concerns for high peak airway pressures and theoretical predictions about regional ventilation distributions.
Hooke's Law
As defined by Hooke's law, stress amplitude varies with the amplitude of deformation and with the material properties of the deformed elastic object. The amplitude of the deformation is commonly expressed as strain. Engineers commonly define strain as a dimensionless ratio (ie, the change in length or volume of a material relative to its unstressed and therefore un-deformed state). However, this definition is not useful in respiratory mechanics because the lung is pre-stressed in situ (ie, exposed to pleural pressure) so that its unstressed volume cannot be known. One may of course deviate from the engineering definition and normalize deformation (ie, VT) by either the volume at end-expiration, or any other volume, such as the volume at zero airway pressure or total lung capacity for that matter.8–10
Needless to say, attempts to define unique strain injury thresholds suffer from this limitation and should be interpreted with caution. The biophysical injury mechanisms associated with large volume oscillations (ie, VT), high end-inspiratory volumes, and low end-expiratory volumes differ from each other. As a result, the interpretation of strain derived from VT divided by end-expiratory volume remains ambiguous. It is therefore preferable to consider these variables in isolation. In fact, by setting limits to VT, PEEP, and Pplat, prevailing ventilator management guidelines do just that.
How Strain-Stress Informs the Interpretation of Clinical Trials and Recent Hypotheses of Ventilator-Induced Lung Injury
As is widely known, a large multi-center clinical trial conducted by the ARDS Network (the ARMA trial) established a survival benefit in subjects with ARDS who were ventilated with low as opposed to high VT (6 mL/kg vs 12 mL/kg predicted body weight).11 Because VT is a critical determinant of strain (regardless of one's chosen definition), this finding is in keeping with biophysical injury mechanisms as they are currently understood.12,13 However, recall that a post hoc analysis that included ARMA trial data suggested that ΔP (and not VT) was the variable that best stratified the biophysical lung injury risk of subjects with ARDS.6 At first glance, this may seem surprising because ΔP and VT are interdependent: ΔP = VT/CRS, where CRS is the compliance of the relaxed respiratory system (the stiffness term in Hooke's law). However, if confirmed, the post hoc analysis suggests that VT-related injury risks vary with CRS.
This hypothesis is attractive for 2 reasons. CRS is a marker of disease severity because CRS varies in proportion to the number of aerated and recruitable lung units (ie, of the size of Gattinoni's baby lung).13 Therefore, a low CRS not only identifies sicker patients with a worse prognosis, but also patients—who by virtue of having fewer recruitable alveoli—are more likely to suffer damage from alveolar overdistention.
A related reason why driving pressure may be superior to VT as initial guide for safe ventilator settings is the fact that normalizing or expressing VT as a function of predicted body weight, as was done in the ARMA trial, fails to take into account the effect of disease on available lung volume. The primary determinants of predictive body weight, namely gender and height, scale with the size of the normal, healthy lung, but not with that of the diseased lung.14
Since the recognition of ventilator-induced lung injury (VILI), researchers and clinicians have sought to identify a singular surrogate that could serve as marker of VILI risk and, therefore, act as a guide for individualizing care. Along these lines, Gattinoni and colleagues have recently proposed that the power (work per unit time) applied by the mechanical ventilator on the relaxed respiratory system may serve that purpose.15 Although it is not overly difficult to measure work on the relaxed respiratory system (based on the area of airway pressure volume loops), there is scant evidence to date in support of this hypothesis.
Skepticism about this hypothesis rests on the uncertainty that contributions of breathing frequency, flow, and VT to power transfer to the lungs may not scale with their respective contribution to injury risk. Moreover, the effects of PEEP on injury risk may not be sufficiently captured by this approach. While there is some evidence that breathing frequency influences certain manifestations of lung injury under specific experimental conditions,7 the many interactions between disease state and breathing frequency, driving pressure, pressure (stress) maxima and minima, and the rate of parenchymal deformation remain undefined.
VILI, Biotrauma, and the Role of PEEP
Even though driving pressure emerged as the single best predictor of biophysical injury risk (in post hoc analyses of large data sets), large pressure changes are unlikely to injure cells and tissues through a single mechanism. That is because mechanical ventilation with large driving pressures also affects vascular barrier properties, surfactant kinetics and function, and amplifies parenchymal stresses attributable to alveolar wall tension and interdependence effects.16
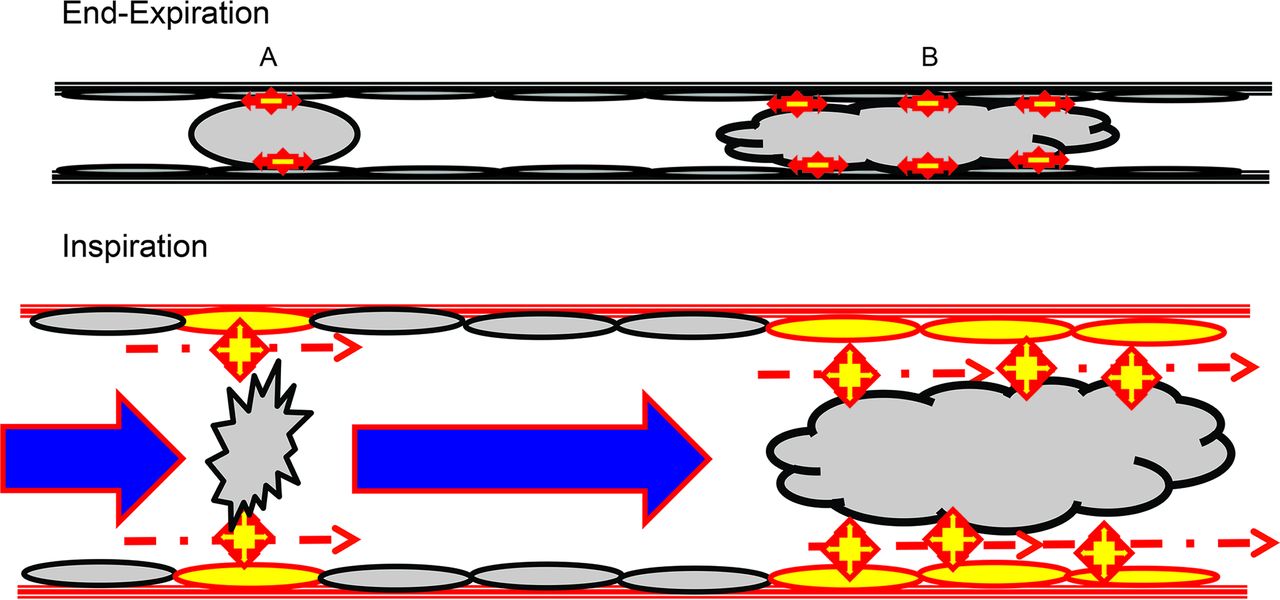
When viewed through the lens of cell and tissue micromechanics, injury manifests as a loss of epithelial and endothelial cell integrity that is accompanied by an influx of inflammatory cells. Stresses that wound epithelial cells tend to be interfacial in nature. They are caused by movement and/or rupture of liquid bridges and liquid plugs that cross the epithelial surface (Fig. 3). Thus, interventions designed to minimize air space fluid and plug formation are generally lung-protective.17

Depiction of shear stress development from the breaking of liquid bridges (A) and liquid plugs (B) formed from pulmonary edema fluid in peripheral airways. The stars represent surface tension forces developed within the foam and plug that are transmitted to the surface of airway epithelium. During inspiration, as the foam menisci are broken and plugs are displaced distally, shear forces develop between the displaced liquid and the relatively stable epithelial cell membranes.
The clinical literature addresses these mechanisms in the context of alveolar recruitment, PEEP management, and the avoidance of atelectrauma. The original description of atelectrauma was based on experiments in unperfused, saline-lavaged rat lungs that were subjected to ex vivo mechanical ventilation. This model underscored the effects of airway and air-space liquid on epithelial integrity and thereby provided a rationale for the lung-protective effects of PEEP and for the salutary effects of fluid restriction in the management of patients with ARDS.18 By increasing end-expiratory lung volume and transalveolar pressure, PEEP reduces the probability of air-space occlusion by liquid bridges and promotes the translocation of edema fluid from air spaces to the interstitium.19
While there is ample evidence that PEEP is lung-protective in patients with ARDS, several large clinical trials have failed to identify a single “best” approach to PEEP management.20–23 Controversies and discordant results may be rooted in less than optimal patient selection criteria, in the choice of procedures and biomarkers used to guide PEEP dosing, and in uncertainty about untoward cardiovascular side effects and their management. The latter concern is true for virtually all clinical trials focused on lung protection and may well explain the apparent harm associated with high-frequency oscillatory mechanical ventilation in a recent study.24
Patients with ARDS are at risk for right heart failure, whereby aggressive recruitment efforts accompanied by high PEEP tend to increase that risk.25 The problem may not be recognized unless the patient becomes hypotensive or is examined using bedside ultrasonography. Faced with a hypotensive patient, the provider may choose to give intravenous fluids, add an inotrope, or reduce PEEP. These decisions are rarely if ever explicitly addressed in standard operating procedures of clinical trials, so it is unclear whether a fall in blood pressure by itself is a sufficiently sensitive or specific guide to appropriate cardiovascular management.
Clinical Management Strategies
In the absence of strong clinical evidence that would favor a specific approach to PEEP management, such management is typically guided by responses in gas-exchange or respiratory-system mechanics. Gas-exchange–based protocols adjust PEEP in relation to FIO2 as originally defined in the standard operating procedure of the ARDSnet trials.11 Compared to the version used in the original ARMA trial (the so-called low PEEP-FIO2 table), a more recent update has adopted a more aggressive recruitment strategy (high PEEP-FIO2 table).22 Protocols based on respiratory mechanics typically adjust PEEP with the goal to minimize ΔP.26 The rationale for this approach is that ventilating patients over the most compliant range of their respiratory system's pressure-volume relationship minimizes the respective injury risks associated with alveolar overdistention and cyclic recruitment.27
There is emerging interest in adjunct technologies including esophageal manometry, lung ultrasonography, and thoracic impedance plethysmography as guides for PEEP management.28,29 While the use of imaging technologies is largely confined to case series and observational studies, a small clinical trial suggested that esophageal-pressure–targeted PEEP management was superior to a gas-exchange–based algorithm.30 Notwithstanding concerns among physiologists and clinicians about the limiting assumptions of esophageal manometry in recumbent ARDS patients, a larger clinical trial that is to address the clinical efficacy of this technique is nearing completion.31
Maximum Lung Stress and Pplat
As already discussed, the injury risk associated with repeated inflations of the lungs and respiratory system to high volumes and pressures depends greatly on the corresponding driving pressure. In the classic experiments of Webb and Tierney,32 rats were ventilated to peak pressures of 40 cm H2O. The lung-protective effect of reducing driving pressures (from 40 to 30 cm H2O) was attributed to a lung-protective effect of PEEP, which had been increased from 0 to 10 cm H2O. It stands to reason, however, that PEEP would not have been (as) protective if driving pressure had not been reduced as well. Needless to say, there is a maximum lung stress that should not be exceeded regardless of the associated driving pressure. Because maximum lung stress (ie, the elastic recoil pressure of the lung at end-inspiration) is rarely measured in clinical practice, most ventilator-management guidelines limit Pplat (ie, the elastic recoil pressure of the relaxed respiratory system at end-inflation) to ≤ 30 cm H2O.
This guideline is based on the assumption that the chest wall mechanical properties are normal so that the corresponding transpulmonary pressure will not exceed 20 cm H2O. Critics of this assumption argue that this estimate was derived from esophageal pressure measurements in seated normal volunteers and therefore does not apply to recumbent patients, particularly those with obesity, ileus, or ascites.33 In such patients, the lung volume at end-expiration is substantially reduced and often requires PEEP settings between 15 and 20 cm H2O simply to counterbalance the abdominal load. Limiting Pplat to ≤ 30 cm H2O under those circumstances would make it impossible provide adequate alveolar ventilation.
Unresolved Issues Surrounding VILI
Despite significant advances in our understanding of the molecular determinants of mechanotransduction, related fundamental research has yet to generate clinically feasible and efficacious interventions targeting VILI risk reduction.34 Experimental VILI models, which generally cannot capture the full scope of patients' disease and health-care exposures, tend to focus on early manifestations of injury such as edema and inflammation. In that context, immunosuppressive effects of pharmacologic and molecular interventions are generally deemed protective, even though it is unclear whether such responses are adaptive or maladaptive. The many unresolved questions surrounding fundamental biophysical mechanisms that shape the responses of injured lungs to deforming stress are beyond the scope of this clinical review.
While attention to VT and driving pressure have contributed to a decline in the incidence of health-care–acquired ARDS, there is a great deal of uncertainty about how to judge VILI risk in a given patient and, therefore, how best to individualize care. For example, some patients with ARDS, when supported in a pressure-pre-set mechanical ventilation mode, choose tidal volumes that exceed lung-protective guidelines. This occurs not infrequently after several days of mechanical ventilation and is thought to reflect the influence of prolonged sedative and narcotic use on respiratory drive and timing.35
In that instance, the provider must question if lung protection still mandates the imposition of low-VT mechanical ventilation, which would require a resumption of sedation and/or neuromuscular blockade, or if the VILI risk has decreased with time from the initial insult. While there are no clinical or experimental data that would speak to this issue, it could be argued that, in the subacute phase of ARDS, damage from interfacial stress is no longer of major concern because air-space fluid will have largely solidified.36 While there is no direct way to measure the mechanical properties of material filling air spaces in injured lungs, one may reasonably postulate that lungs filled with liquid are more recruitable than consolidated lungs.
Uncertainty about patient-specific VILI risk factors also dominates debates about indications and timing of intubation and invasive mechanical ventilation, and about the use of rescue therapies such as extracorporeal membrane oxygenation (ECMO) and extracorporeal CO2 removal. While much of the VILI literature has focused on mechanical ventilation with high VT and low PEEP, there is reason to believe that spontaneous breathing would also produce injury as long as lung deformations are comparable between the modes of breathing.37
There is an emerging appreciation that excessive oxygen supplementation contributes to and amplifies lung deformation injury.38,39 Nevertheless, there is a great deal of uncertainty how to individualize O2 dosing. While most oxygen-titration guidelines target arterial oxygen saturations between 88% and 92%, it is unclear if and how these targets should be modified in patients with metabolic or cardiovascular stress. Regardless, prolonged exposure (ie, days) to FIO2 ≥ 0.80 should be avoided and a safer goal would be a target of FIO2 ≤ 0.60.39 Furthermore, there is no consensus on the relative risks and benefits of hypercapnia.40 Given the topographical heterogeneity of ventilation-perfusion ratios in patients with ARDS, there must be a corresponding heterogeneity in alveolar gas tensions and local pH. Their effects on mechanotransduction and local immune responses remain largely unexplored.
Practical Bedside Strategy for Implementing Lung-Protective Ventilation in Severe ARDS
Based upon the above discussion of VILI and the profoundly altered chest mechanics in ARDS, the following algorithm can be applied specifically to patients with particularly severe presentations who are unstable and warrant hyperoxygenation, sedation, and neuromuscular blockade in conjunction with intubation (Fig. 4).
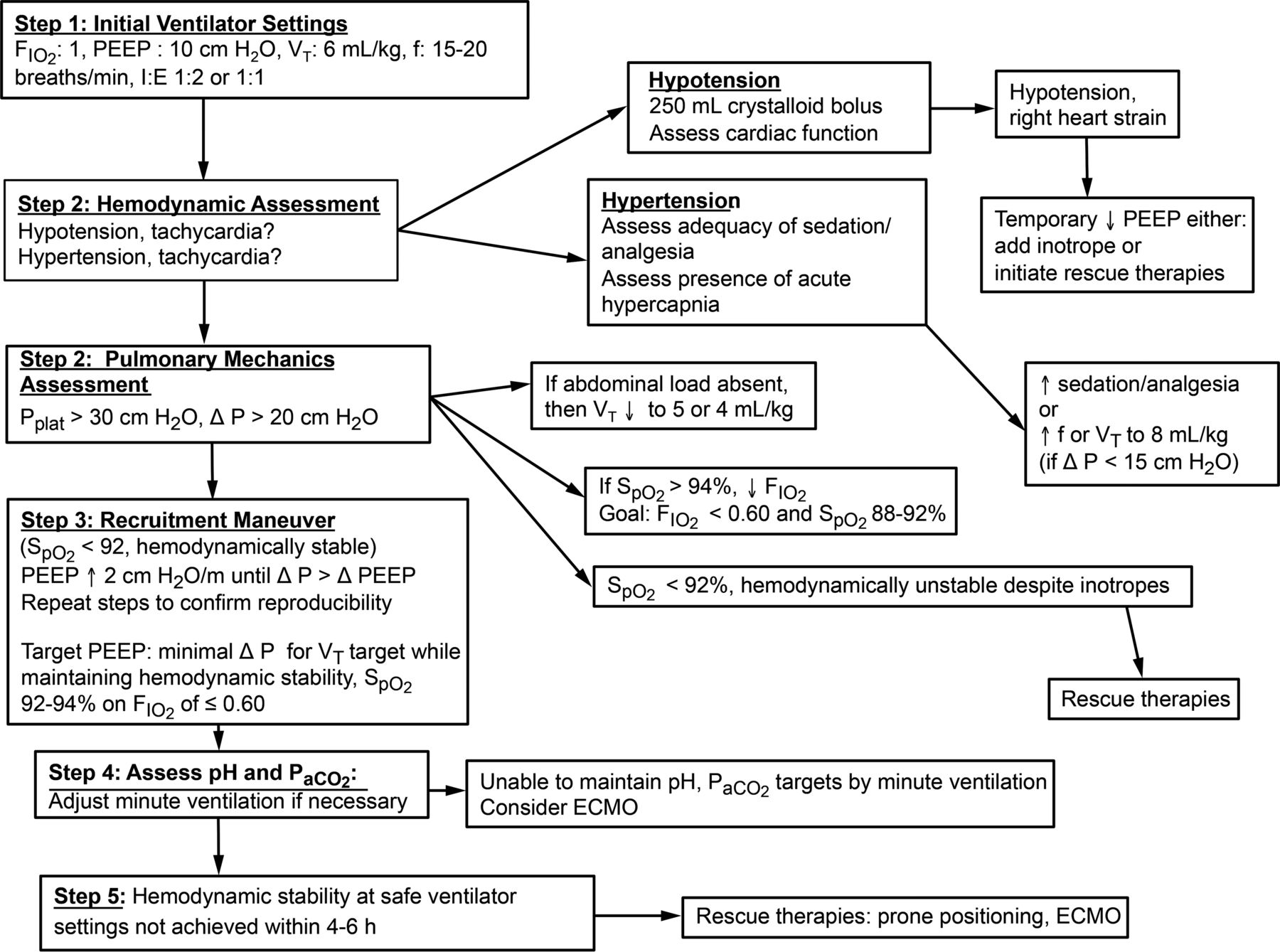
Schematic representation of a pragmatic guide to safer ventilator management of unstable patients with severe lung injury. ECMO = extracorporeal membrane oxygenation, Pplat = plateau pressure.
Step #1
Set FIO2 to 1.0, PEEP to 10 cm H2O and initiate volume-pre-set mechanical ventilation with VT = 6 mL/kg predicted body weight at a frequency of 15–20 breaths/min and an inspiratory flow setting between 0.75 and 1 L/s (targeting an initial I-E ratio between 1:2 and 1:1).
Rationale.
Using a volume-pre-set mode with constant (square wave) inspiratory flow as an initial approach facilitates the assessment of respiratory mechanics (particularly the contribution of airways resistance) and recruitment effects in that mode.
Step #2
Assess blood pressure (invasive measurement preferred), heart rate, Pplat, ΔP, and arterial oxygen saturation via pulse oximetry (SpO2).
a) If these variables are within tolerance limits, proceed to Step #3.
b) If the patient has tachycardia, hypotension, and/or has substantial pulse-pressure variations, give an intravenous fluid bolus (eg, an initial 250 mL bolus of a balanced electrolyte solution) and assess cardiac function using a portable echocardiograph/ultrasound probe. If the exam shows right heart strain in a hypotensive patient, temporarily reduce PEEP, consider an inotrope, and initiate a rescue therapy (typically prone posture and/or ECMO).
c) If the patient is hypertensive, assess the level of sedation and evaluate for undertreated/unrecognized pain. Afterward, consideration can be given to raising minute volume as hypertension could be a response to acute hypercapnia. Raise minute volume either by increasing the frequency (30–35 breaths/min) or VT in steps to 7–8 mL/kg predicted body weight (particularly if ΔP is < 15 cm H2O), or some combination thereof.
d) If Pplat is >30 cm H2O, indicating that the corresponding driving ΔP is >20 cm H2O, reduce VT in steps to 4–5 mL/kg predicted body weight. In patients with large abdominal loads, exclude a spurious elevation of ΔP on account of expiratory flow limitation, airway closure, and inadvertent PEEP.
e) If SpO2 is < 92% in the presence of hemodynamic stability, initiate recruitment efforts (as outlined in Step #3). If SpO2 is <92% in a hemodynamically unstable patient who fails to correct hypoxemia in response to hemodynamic support measures, proceed to rescue therapies.
f) If SpO2 is > 94%, reduce FIO2 with the goal to at least temporarily target a FIO2 of < 0.80 and an SpO2 of 88–92%, and then proceed with recruitment efforts.
Step #3
Increase PEEP by 2 cm H2O and monitor the SpO2, Pplat, ΔP, and hemodynamic response. Unless blood pressure falls, repeat PEEP increments every 30–60 s until ΔP starts to rise by ≥ 2 cm H2O (ie, the change in Pplat exceeds the change in PEEP by ≥ 2 cm H2O). Such a rise suggests that the lung is approaching its maximum volume, at which point further recruitment attempts could exceed parenchymal stress tolerance. Then reduce PEEP in increments of 2 cm H2O until ΔP returns to its minimum level.
Repeat this process several times until the response becomes reproducible. At that point, any PEEP setting compatible with minimum driving pressure (for a given VT) may be chosen. Some practitioners may decide to choose the highest PEEP at which this condition is met, whereas others may choose the lowest corresponding value in patients with limited cardiovascular reserve.
Rationale.
As already discussed, there are many acceptable ways to arrive at an acceptable PEEP setting. The approach suggested here is consistent with the stress-index philosophy and should facilitate further reductions in FIO2.
Step #4
Check arterial blood gases primarily to assess PaCO2 and pH and adjust minute volumes accordingly. If that cannot be accomplished safely, consider ECMO or extracorporeal CO2 removal.
Step #5
If the desired outcome (ie, hemodynamic stability at safe ventilator settings and FIO2 ≤ 0.60) cannot be achieved within 4–6 h, consider rescue therapies, foremost prone positioning and depending on availability ECMO.
Footnotes
- Correspondence: Richard H Kallet MSc RRT FAARC, Department of Anesthesia and Perioperative Care, University of California, San Francisco at Zuckerberg San Francisco General Hospital, Building 5:GA-2, 1001 Potrero Ave, San Francisco, CA 94110. E-mail: rich.kallet{at}ucsf.edu.
Dr Hubmayr discloses relationships with Philips Research North America and Global Blood Therapeutics. Mr Kallet discloses relationships with Nihon Kohden, Getinge Group USA, and Philips Respironics.
- Copyright © 2018 by Daedalus Enterprises





{kind=link}
{kind=link}
{kind=link}
{kind=link}