

Introduction
Albuterol is an uncommonly recognized culprit in causing lactic acidosis. While treating asthmatics, lactic acidosis creates a paradoxical situation in which, despite improvement in bronchospasm, patients look more dyspneic as a compensatory mechanism for metabolic acidosis. This might lead to misinterpretation of the situation with increased albuterol dosing, creating a sort of vicious cycle ultimately resulting in respiratory failure. Healthcare providers treating asthmatics should be well aware of this entity, so that they can adjust therapy accordingly.
Case Summary
A 22-year-old African American female with history of asthma presented to the emergency department with the chief complaint of shortness of breath. Per the patient, this was precipitated by an upper respiratory infection. Her breathing progressively got worse over a couple of days, and there was no relief even with increased use of albuterol. She also complained of wheezing, chest tightness, and frequent awakenings at night. The patient had asthma since childhood, with an episode of intubation. Overall, her asthma was well controlled, and the last “bad” attack was 3 years ago. She had substantial seasonal allergies, and her asthma triggers include pollen, dust, perfumes, and weather changes. She never smoked, and denied having any pets. She had a family history of asthma in her mother and siblings. She was taking albuterol, fluticasone/salmeterol 250/50 μg, and montelukast 10 mg. The rest of the review of systems was unremarkable.
Physical examination showed a well built hypertensive (blood pressure 160/74 mm Hg), tachycardic (heart rate 140 beats/min) and tachypneic (respiratory rate 24 breaths/min) patient in distress. She was able to speak in full sentences, and was saturating 100% on 2 L/min of oxygen. Her best peak flow documented was 300 L/min, and she did only 120 L/min. She had substantial bilateral expiratory wheezing on examination. The rest of her physical examination was unremarkable, and her chest x-ray was normal. Her arterial blood gas values on arrival showed pH 7.33, PaCO2 43 mm Hg, PaO2 180 mm Hg, bicarbonate 22 mEq/L, saturation 99%, and lactate 3 mEq/L, on 2 L/min of oxygen. Her labs were normal, including a negative toxic drug screen and normal liver function tests. The patient received prednisone 60 mg and a total of 5 albuterol nebulizations (2.5 mg each) in the emergency room. After these treatments, despite improvement in her peak flow to 150 L/min and decreased wheezing, she became more dyspneic. A repeat arterial blood gas then showed pH 7.33, PaCO2 35 mm Hg, PaO2 90 mm Hg, bicarbonate 18 mEq/L, saturation 98%, and lactate 6.8 mEq/L, on 2 L/min of oxygen.
The ICU was consulted for worsening lactic acidosis and respiratory distress. In the absence of hypoxia or global tissue hypoperfusion, with normal liver function tests and a negative toxic drug screen, albuterol was suspected as the cause for her lactic acidosis. Albuterol was discontinued, the patient was managed with systemic steroids and ipratropium, and the lactic acidosis resolved within the next 24 hours. The arterial blood gas analysis next day showed pH 7.43, PaCO2 40 mm Hg, PaO2 80 mm Hg, bicarbonate 27 mEq/L, saturation 99%, and lactate 1.2 mEq/L, on 2 L/min of oxygen, and peak flow improved to 250 L/min. She was eventually discharged home in a stable condition.
Discussion
Lactic acid is the end product of anaerobic metabolism of glucose. At times it is also the only source of energy during anaerobic metabolism.1 In fact, reduction of pyruvate to lactate by lactate dehydrogenase enzyme is the only mechanism through which lactic acid is produced in the body:
There are 2 main mechanisms of lactate production. Woods and Cohen classically described them as type A and type B lactic acidosis. In the familiar type A lactic acidosis, lactic acid is produced in response to a hypoxic or hypoperfusion state. Here, in the absence of oxygen, pyruvate dehydrogenase enzyme is inhibited, preventing conversion of pyruvate to acetyl-CoA. This forces pyruvate to undergo anaerobic metabolism to lactate rather than entering the citric acid cycle. Here the lactate/pyruvate ratio is increased. Type B lactic acidosis, on the other hand, is generated in the absence of hypoxia or hypoperfusion. The common causes of type B lactic acidosis are drugs, malignancies, and inborn errors of metabolism2 (Table).
Causes of Lactic Acidosis
With regards to asthma, the most common metabolic disturbances seen during an acute attack are respiratory alkalosis, followed by respiratory acidosis as fatigue sets in.3 Type A lactic acidosis is also seen during asthma exacerbation, due to hypoxemia. Pulsus paradoxus and intrinsic PEEP decrease cardiac output and venous return, respectively, leading to type A lactic acidosis. In addition, type B lactic acidosis has also been historically described in asthmatics.
Several mechanisms have been proposed to explain the type B lactic acidosis in asthmatics. Respiratory alkalosis during asthma attack has been shown in animal studies to stimulate phosphofructokinase, the rate limiting step in glycolysis. This leads to enhanced glycolysis, more pyruvate, and ultimately more lactate production.4,5 However, lactic acidosis through this mechanism is very mild in humans and occurs only at extreme alkalosis (pH > 7.6 and PaCO2 < 20 mm Hg).6 Another important mechanism proposed is that lactic acidosis occurs secondary to increased respiratory muscle work load against a closed upper airway, leading to increased oxygen demands and lactic acidosis.7 Interestingly, lactic acidosis was shown to occur in mechanically ventilated and paralyzed asthmatics, where respiratory muscle fatigue should not be an issue.8–10 A mechanism of relative hepatic ischemia and impaired lactate clearance due to venous congestion from intrinsic PEEP was also proposed.11
With no evidence supporting the above mentioned explanations, a mechanism of hyperadrenergic state was suggested. It was thought that lactic acidosis in asthmatics occurs secondary to an endogenous or exogenous hyperadrenergic state. This could be either from stress and anxiety associated with an asthma attack, or from drugs given during the attack. One such exogenous agent well described to induce lactic acidosis is albuterol.12 Albuterol is a β2 agonist used for bronchodilation in asthma. Albuterol causes lactic acidosis by a combination of factors, but the exact etiology and why only a proportion of patients develop this condition remain to be elucidated. Albuterol produces lactic acidosis through its metabolic effects. By creating a hyperadrengic state it enhances glycogenolysis and gluconeogenesis, leading to more glucose, enhanced glycolysis, and pyruvate production. At the same time, enhanced lipolysis and increased free fatty acids inhibit pyruvate dehydrogenase enzyme, preventing pyruvate from entering the Krebs cycle. This causes pyruvate reduction to lactate12,13 (Fig. 1). Also, simultaneous use of steroids during an asthma attack enhances the β2 receptor sensitivity, further potentiating lactate production.9

Albuterol induced lactic acidosis.
Albuterol-induced lactic acidosis is an uncommonly recognized culprit in lactic acidosis, although it has been well described in the literature. Ritodrine infusion, a β2 agonist used to prevent premature labor in obstetric patients, was shown to produce lactic acidosis.14,15 β2 agonist-induced lactic acidosis was described in healthy volunteers,16 and also in asthmatics by both intravenous17–19 and inhalation routes.10,20–22
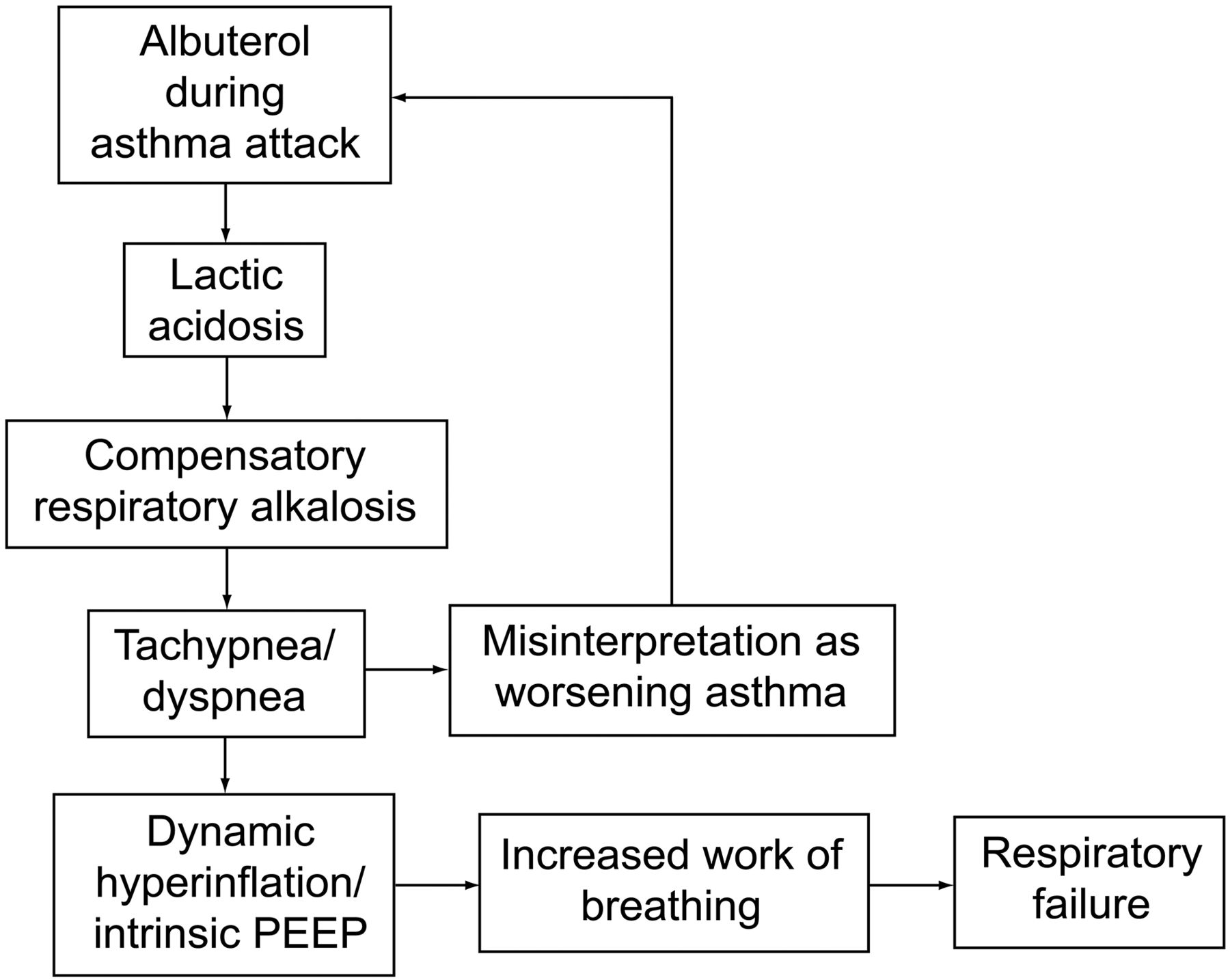
The clinical importance of this is that albuterol-induced lactic acidosis creates a paradoxical situation. During asthma exacerbations, after treatment with albuterol, in spite of bronchodilation, patients look more dyspneic and tachypneic, as they are compensating for lactic acidosis. Healthcare providers might misinterpret this situation as worsening respiratory failure and give more albuterol, creating a vicious cycle and ultimately leading to respiratory failure18 (Fig. 2). The best way to identify this situation is with serial peak flow measurements and physical examination. There is no clear evidence-based management strategy for this paradoxical situation. Historically, different strategies, like continuing the albuterol at the same dose, decreasing the dose, or changing over to ipratropium, were used. Further studies are needed to establish the exact epidemiology, etiology, and management of this unique paradoxical situation.
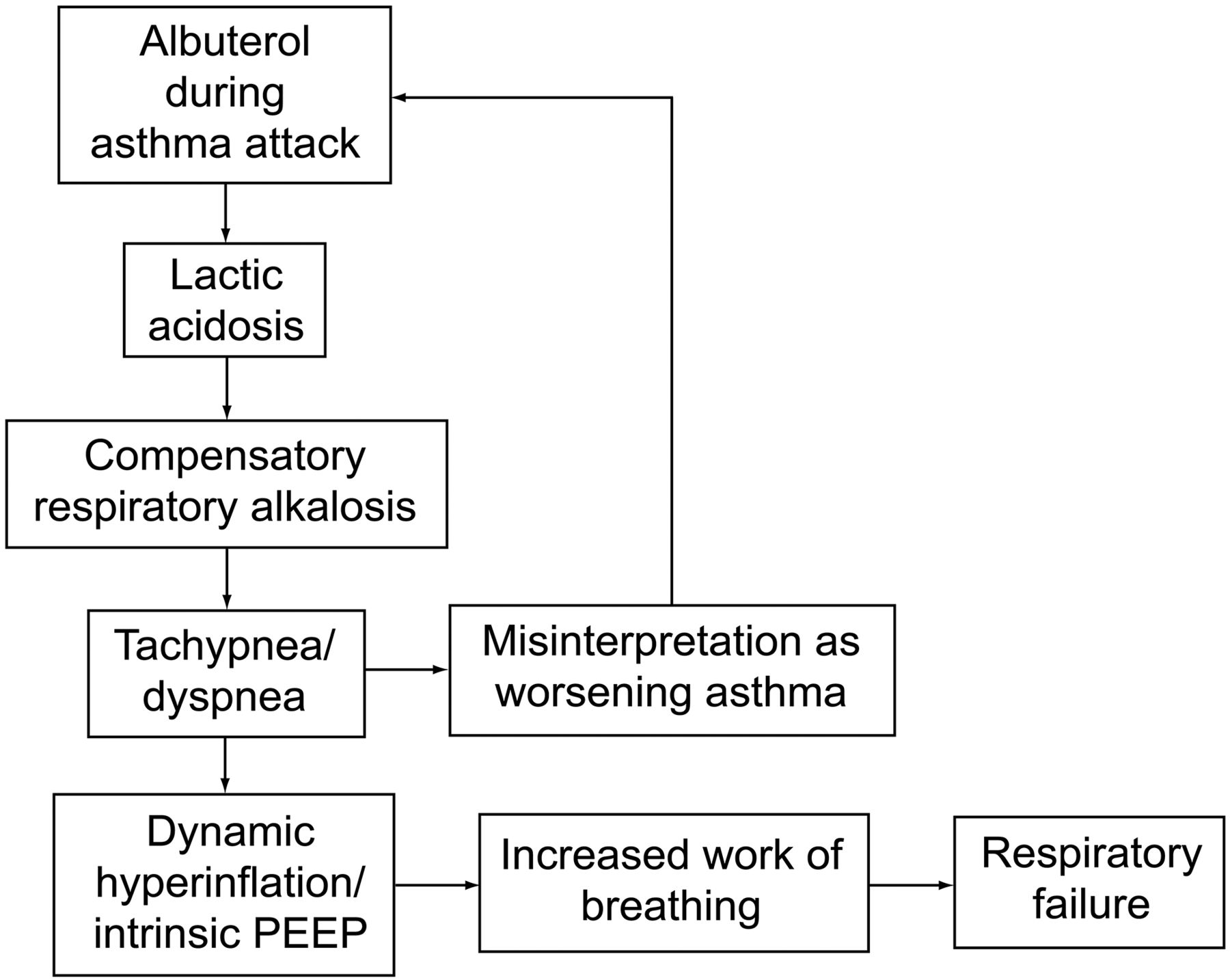
Albuterol related paradoxical situation.
So why did we blame albuterol for our patient's lactic acidosis? Our patient had no evidence of hypoxia or global tissue hypoperfusion. She had normal liver function tests and a negative toxic drug screen. Lactic acidosis developed after aggressive albuterol treatments, and it resolved once these treatments were discontinued. Historically, albuterol-induced lactic acidosis has been well described. So, by deduction, albuterol was assumed to be the culprit. Although a component of lactic acidosis secondary to endogenous hyperadrenergic state may exist, it is hard to delineate it from albuterol-induced lactic acidosis.
Teaching Points
Continuous clinical assessment and close monitoring of peak flow is crucial while managing asthmatics.
Intense use of β2 agonists may cause lactic acidosis through their metabolic effects.
Any discrepancy between worsening of dyspnea and resolution of bronchospasm, with no other explanation for lactic acidosis, should lead the healthcare provider to consider albuterol as the etiology.
Respiratory compensation may be misinterpreted, leading to unnecessary intensification of management and ultimately resulting in respiratory failure.
Footnotes
- Correspondence: Venkata R Dodda MD, Department of Pulmonary Medicine, Harlem Hospital, 506 Lenox Avenue, New York NY 10037. E-mail: ranga_dr{at}yahoo.com.
Dr Dodda presented a version of this paper at the Chest 2011 conference, held October 22–26, in Honolulu, Hawaii.
The authors have disclosed no conflicts of interest.
- Copyright © 2012 by Daedalus Enterprises Inc.





{kind=link}
{kind=link}