

Abstract
BACKGROUND: Between 2012 and 2017, 25 new medications or combination products were approved by the Food and Drug Administration (FDA) for use in treatment of chronic lower respiratory diseases (CLRDs). With limited data on post-marketing patient exposure to these drugs, their safety profiles remain unknown. This study aims to provide post-marketing surveillance of these medications.
METHODS: A list of new CLRD medications approved between 2012 and 2017 was generated through searches on Drugs.com (https://www.drugs.com), FDA.gov (https://www.fda.gov), and IBM Micromedex (https://www.micromedexsolutions.com/home/dispatch/ssl/true). Data describing adverse drug reactions (ADRs) were collected from the FDA Adverse Event Reporting System for analysis. Of the 25 identified medications, we selected 4 medications indicated for asthma or COPD with at least 500 reports. Only ADRs catalogued with these medications as the primary suspect were analyzed. Reporting odds ratios were calculated for the top 10 ADRs of each CLRD medication.
RESULTS: A total of 61,682 ADR reports were collected for newly approved CLRD medications (n = 27,190 older adults; n = 30,502 male). Reports of COPD medications (umeclidinium and umeclidinium/vilanterol) indicate that umeclidinium/vilanterol yielded a higher reporting odds ratio than umeclidinium alone for reports of pain. Fluticasone furoate/vilanterol had higher reporting odds ratios for cough, pain, and dizziness than budesonide/formoterol and fluticasone propionate/salmeterol.
CONCLUSIONS: Our findings suggest that the incidence of different adverse events experienced by patients in post-marketing reports resembles the incidence reported in pre-marketing clinical trials for COPD medications, except for fluticasone furoate/vilanterol, which has several differences.
- pharmacovigilance
- post-marketing
- chronic obstructive pulmonary disease
- asthma
- reporting odds ratio
- adverse drug reaction
Introduction
Chronic lower respiratory diseases (CLRDs) encompass a variety of disorders affecting the lungs and other parts of the respiratory system. The World Health Organization defines CLRDs as diseases of the airways and other structures of the lung.1 The term encompasses COPD, asthma, occupational lung diseases, and pulmonary hypertension. CLRDs are not curable, and treatment generally focuses on symptom management.
CLRD was the fourth-leading cause of death in the United States in 2017, with 160,201 deaths attributed.2 Worldwide, an estimated 65 million people suffer from COPD and 334 million suffer from asthma. Asthma is the most common chronic disease in childhood and affects an estimated 14% of all children globally.3 In the United States, asthma alone cost the health care system around $82 billion in 2013, with the average per-person incremental prescription cost of $1,830 and an average per-person incremental hospitalization cost of $529 per year.4 In 2010, COPD cost the United States health care system $32.1 billion, and annual costs are projected to grow to $49.1 billion by the end of 2020.5 The prevalence of CRLDs rises each year and may be driven by factors associated with an ever-growing global urban population, such as pollution from automobile exhaust and industrial processes.6
Medications used in the management of CLRDs, such as the short-acting β agonist albuterol, are well-established in current practice. Between 2012 and 2017, 25 new medications, including combination products of new and old medications, were approved by the Food and Drug Administration (FDA) for use in CLRD. As with other newly marketed medications, data regarding patient exposure to these drugs are limited. As such, the full safety profiles of new CLRD medications are unknown. Post-marketing surveillance and pharmacovigilance analyses can address gaps between safety profile information gained from controlled clinical trials and real-world patient experience. Identifying and minimizing these gaps will not only allow patients and health professionals to make appropriate treatment choices but also minimize wasted health expenditures.
Post-marketing surveillance involves monitoring the safety of a drug after it is approved for patient use. Post-marketing surveillance can identify new adverse drug reactions (ADRs) that were not observed in controlled clinical trials, further confirm the prevalence of established ADRs, and reveal new beneficial effects not investigated during the drug development process.7 The FDA Adverse Event Reporting System (FAERS) is a vital database for post-marketing surveillance operations. Anyone may report suspected ADRs to the database and include information relating to the suspected drug, the patient, and the ADR(s) in question. This study aims to provide a post-marketing surveillance of the 25 CLRD medications that were approved between 2012 and 2017 using FAERS data.
Quick Look
Current Knowledge
Between 2012 and 2017, a total of 25 new medications or combination products were approved by the FDA with indications for chronic lower respiratory disease. Information on the safety profiles of these agents is limited to results of clinical trials, and post-marketing data are needed for a comprehensive understanding.
What This Paper Contributes to Our Knowledge
The number of voluntary adverse event reports resembled much of the existing adverse event profiles for umeclidinium, umeclidinium/vilanterol, and roflumilast, whereas reports for fluticasone furoate/vilanterol showed safety signals inconsistent with existing adverse profiles and require further investigation. Respiratory therapists, as patient care advocates, can contribute to the detection of possible adverse drug reactions to provide early management when possible.
Methods
Study Design
This was a post-marketing pharmacovigilance analysis focused on ADRs reported in FAERs for CLRD medications that were approved between 2012 and 2017. Institutional review board approval was not required because the study used de-identified public data.
Inclusion and Exclusion Criteria
A list of CLRD medications used for this analysis was generated through searches on Drugs.com (https://www.drugs.com, Accessed May 12, 2021), FDA.gov (https://www.fda.gov, Accessed May 12, 2021), and IBM Micromedex (https://www.micromedexsolutions.com/home/dispatch/ssl/true, Accessed May 12, 2021). Medications had to be approved by the FDA between 2012 and 2017 to be included in this study. Information obtained about the medications include their brand and generic names, indication, disease state, approval year, all reported ADRs, and suspected medication for their ADRs categorized as the primary, secondary, or tertiary suspect. Between 2012 and 2017, a total of 25 new medications with at least one indication for CLRD were approved by the FDA. This study focuses on those drugs approved for asthma or COPD. FAERS reports were included if the primary suspect drug was one of the drugs of interest and if the patient’s age was > 18 y. Of these, we focused on 4 medications with ≥ 500 reports to ensure an adequate sample size for analysis.8
An ADR was defined as an appreciably harmful or unpleasant reaction resulting from an intervention related to the use of a medicinal product.9 Reports such as “drug dispensing error,” “drug dose omission,” and “product issues” were excluded from analysis because these do not accurately represent ADRs but rather product or administration errors. “Nontherapeutic responses” was included as an ADR to define “drug ineffective” and “drug intolerance” in our data.
Classification of Drugs and ADRs
CLRD medications were classified by FDA-approved indication into 4 categories: asthma, COPD, idiopathic pulmonary fibrosis, and seasonal allergic rhinitis. This study focuses on medications approved for asthma or COPD. Asthma is a chronic inflammatory disorder involving the airways, where many cells and cellular elements such as mast cells, eosinophils, T lymphocytes, macrophages, neutrophils, and epithelial cells play an important role.10 COPD is a group of chronic diseases that cause breathing-related problems from air flow blockage.11
Two older medications, budesonide/formoterol (BUD/FORM) and fluticasone propionate/salmeterol (FP/SAL), approved before 2012, were chosen for comparison with the newer agent fluticasone furoate/vilanterol (FF/VI). BUD/FORM and FP/SAL were selected because they are in the same class and share the same indications with FF/VI. FF, BUD, and FP are all corticosteroids, whereas VI, FORM, and SAL are all long-acting β agonists. FF/VI, BUD/FORM, and FP/SAL are all approved for use in asthma and COPD.12-14
Data Collection
The open-source Project Aeolus (https://project-aeolus.com, Accessed May 12, 2021) was used to represent FAERS data.15 FAERS medication names were mapped to RxNorm standard code ingredients (https://www.nlm.nih.gov/research/umls/rxnorm/index.html, Accessed May 12, 2021) and clinical medication forms for multi-ingredient medications using the Observational Health Data Science and Informatics (OHDSI) Athena Vocabularies and the Usagi mapping tool (https://www.ohdsi.org/analytic-tools/usagi, Accessed May 12, 2021). Additional candidate mappings were created by matching ingredients and clinical medication names to FAERS medication names using a Levenshtein distance of 1. Two investigators (CP, HK) independently reviewed the candidate matches and re-solved differences via consensus. FAERS medication indications and reactions were similarly mapped from the Medical Dictionary for Regulatory Activities (MedDRA) preferred term to Systematized Nomen-clature of Medicine - Clinical Terms standard codes using the OHDSI Athena Vocabularies.
Reports for respiratory medications included in the study were collated for analysis from Quarter 1, 2012, through Quarter 1, 2019, to allow for a 2-y buffer in ADR reports after availability on the market. Reports included in the analysis were narrowed to respiratory medications catalogued as the primary suspect in the FAERS dataset to only incorporate ADRs reported for our drugs of interest. Similar ADRs were consolidated to a common, higher MedDRA level, the Higher-Level Term for medications included in the statistical analysis. Data on patient and reporter demographics were also obtained.
Statistical Analysis
Patient and reporter demographics were analyzed using descriptive statistics. The top 10 ADRs of all 7 CLRD medications were identified with StataSE 16.1 (StataCorp, College Station, Texas). Reporting odds ratios were calculated for the top 10 ADRs of each CLRD medication using the formula 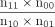 , where n11 is the number of reports for interested ADR from interested medications, n10 is the number of reports for interested ADR from non-interested medications, n01 is the number of reports for every other ADR from interested medications, and n00 is the number of reports for every other ADRs from non-interested medications. ADRs were included in the results and in supplementary tables for analysis only if their reporting odds ratio was significant (see the supplementary materials at http://www.rcjournal.com).
, where n11 is the number of reports for interested ADR from interested medications, n10 is the number of reports for interested ADR from non-interested medications, n01 is the number of reports for every other ADR from interested medications, and n00 is the number of reports for every other ADRs from non-interested medications. ADRs were included in the results and in supplementary tables for analysis only if their reporting odds ratio was significant (see the supplementary materials at http://www.rcjournal.com).
Results
Demographics
A total of 7,952,147 ADR reports were reported in the FAERS database between Quarter 1, 2012, and Quarter 1, 2019, with 61,682 reports involving a newly approved CLRD medications (0.81%). Both patient and reporter demographics are displayed in Table 1. Missing data demonstrate that 96.6% of all observations had data on sex, 57.9% on age, and 98.6% on reporter occupation (see the supplementary materials at http://www.rcjournal.com). Supplemental materials include the 10 most frequently reported ADRs for total medications, and the 10 most frequently reported ADRs of each medication to highlight the general trend of ADRs in CLRD medications (see the supplementary materials at http://www.rcjournal.com). More than three-quarters of patients with reports were > 65 y old. The ratio of males to females was almost evenly split. Eighty percent of reports were from the United States.
Patient and Reporter Demographics in Adverse Events Reports
Approved for COPD Only: UMEC, UMEC/VI, Roflumilast
A total of 16,425 ADRs were reported for the 2 dry-powder inhalers approved for COPD: umeclidinium/vilanterol (UMEC/VI) and umeclidinium (UMEC). Table 2 shows that UMEC had a greater reporting odds ratio compared to UMEC/VI for pain (1.48 [95% CI 1.13–1.93]). The supplemental materials include sex and age analyses of these medications (see the supplementary materials at http://www.rcjournal.com). Female patients had a greater reporting odds ratio compared to male patients for nontherapeutic responses (1.27 [95% CI 1.04–1.56]), while male patients had a greater reporting odds ratio for cough (1.50 [95% CI 1.20–1.87]). The reporting odds ratio of older adults was greater than that for young adults for nontherapeutic responses (1.45 [95% CI 1.02–2.08]).
Reporting Odds Ratios for Adverse Drug Reactions Dry Powder Inhalers Approved Between Q1 2012 and Q1 2017
A total of 2,064 ADRs were reported for roflumilast. Female patients had a greater reporting odds ratio compared to male patients for nausea and vomiting (1.99 [95% CI 1.34–2.96]) and pain (2.02 [95% CI 1.32–3.09]) (see the supplemental materials at http://www.rcjournal.com).
Approved for Both Asthma and COPD: FF/VI
A total of 55,079 ADRs were reported for budesonide/formoterol (BUD/FORM) (n = 47,925) and FF/VI combined (n = 7,154) (see the supplemental materials at http://www.rcjournal.com). Table 3 indicates that FF/VI had a greater reporting odds ratio compared to BUD/FORM for cough (2.99 [95% CI 2.64–3.38]), dysphonia (2.75 [95% CI 2.33–3.26]), pain (6.38 [95% CI 5.23–7.78]), and dizziness (2.56 [95% CI 2.02–3.24]).
Reporting Odds Ratios for Adverse Drug Reactions of BUD/FORM* and FF/VI†
A total of 37,622 ADRs were reported for fluticasone propionate/salmeterol (FP/SAL) (n = 30,468) and FF/VI (n = 7,154) combined (see the supplemental materials at http://www.rcjournal.com). Table 4 shows that FF/VI had a greater reporting odds ratio compared to FP/SAL for dyspnea (2.54 [95% CI 2.25–2.87]), cough (3.60 [95% CI 3.06–4.23]), pain (2.45 [95% CI 2.05–2.92]), and dizziness (1.78 [95% CI 1.40–2.25]).
Reporting Odds Ratios for Adverse Drug Reactions of FP/SAL* and FF/VI†
Discussion
In this retrospective pharmacovigilance study, 25 recently approved CLRD medications were reviewed using FAERS data. Of these medications, 4 were chosen for analysis: UMEC, UMEC/VI, roflumilast, and FF/VI. To our knowledge, this is the first comprehensive evaluation of post-marketing surveillance of new CLRD products. Our results suggest that COPD medications (ie, UMEC, UMEC/VI, and roflumilast) have a post-marketing safety profile similar to that reported in their clinical studies, whereas FF/VI seemed to have novel ADRs not fully captured in its product labeling. There were some gender and age differences with adverse event reports found in our data. These patient safety findings are important for respiratory therapists, as patient care team members, to understand and contribute to possible ADR detection on hospital admission or during in-patient care, thus leading to early management when possible.
Of the total reporters in our data, 70% were consumers. The large percentage of consumer reporters likely stems from the nature of respiratory disease medication administration, as most are self-administered at home without direct oversight from a health care professional. The FAERS database is therefore a valuable resource for obtaining patient-centered reports for CLRD treatments.
Both UMEC inhaler powder and UMEC/VI inhaler powder received approval in 2013 for patients with COPD. Because both medications include UMEC in their ingredients, their safety profiles are prone to be very similar. A 52-week, double-blind, placebo-controlled, parallel-group safety trial of 563 subjects was conducted to compare safety and tolerability of UMEC and UMEC/VI. Various types of infection were observed to be the most common ADRs in both groups, followed by headache, back pain, and hypertension.16 This similarity is also present in our data (see the supplemental materials http://www.rcjournal.com). Excluding medication administration errors, the most reported ADRs for both UMEC and UMEC/VI were dyspnea, cough, pain, and asthenic condition. Because many of these are patient-reported ADRs, it is possible that reports of infections are unlikely while the symptoms of various infections are provided. It is interesting, however, to see that UMEC had a higher reporting odds ratio of pain than UMEC/VI in our findings (Table 2). This is contrary to the EMAX trial (Early Maximization of bronchodilation for improving COPD stability) that was conducted for 24 weeks to compare the efficacy and safety of UMEC and UMEC/VI.17 In the published trial, about 2% and 1% of subjects in UMEC/VI group reported pain in extremity and the neck, respectively. However, < 1% of the reports in UMEC group were for pain.17 It is difficult to conclude that the clinical study yielded false safety outcomes because there is an innate bias in the FAERS data that comes with the nature of voluntary reporting. However, the higher risk of patients on UMEC feeling pain in this real-world data may raise an important awareness for clinicians when they make patient-specific decisions on determining the ideal COPD treatment. Further post-marketing surveillance should be continued to strengthen and establish actual pain rates in both medications.
Roflumilast is an oral tablet indicated for a treatment to reduce the risk of COPD exacerbations in patients with severe COPD associated with chronic bronchitis and history of exacerbations.18 A post hoc, pooled analysis of 2 pre-marketing clinical studies of roflumilast revealed that the most common ADRs reported in the roflumilast group were diarrhea (12.1%), nausea (6%), and weight loss (7.5%).19 The roflumilast package insert also suggests that diarrhea and nausea were the 2 most common ADRs that led to discontinuation in all clinical trials conducted during drug development.19 A retrospective real-world study was conducted for 3 y post-marketing in subjects using roflumilast. Although limited with a small sample size of 83, the findings indicate that weight loss (10.8%) was the most common ADR, followed by loss of appetite (10.8%) and nausea (8.4%).20 These findings in pre-marketing and post-marketing studies align with our post-marketing findings using FAERS data. The supplemental materials indicate that diarrhea (7.1%), nausea and vomiting symptoms (5.5%), pain (5.0%), and weight loss (4.7%) were the 4 most reported ADRs for roflumilast. Because our findings with patient-reported data from FAERS resemble the safety profile of roflumilast extracted from various clinical studies, it is important to closely monitor and consider patients’ tolerance of roflumilast.
FF/VI inhalation powder was approved by the FDA in 2013 for maintenance treatment of COPD or asthma in patients > 18 y old.13 FF/VI is a combination product of a corticosteroid (FF) and a long-acting β2-adrenergic agonist (VI), just like the preexisting medications BUD/FORM and FP/SAL. BUD/FORM and FP/SAL were approved by the FDA in 2006 and 2000, respectively, therefore many clinicians are familiar with patient responses to these medications. Their safety profile is also being updated actively post-marketing. For example, in 2017, the FDA removed the boxed warning for serious asthma-related outcomes from the BUD/FORM drug label after sufficient evidence was evaluated post-marketing.21 On the contrary, FF/VI has not established a strong safety profile post-marketing because it is relatively new in the market. The primary aim for comparing FAERS data between FF/VI and the 2 preexisting medications is to raise awareness to possible adverse events that require monitoring.
The supplemental materials show that BUD/FORM had > 6 times the total number of ADRs reported than FF/VI (47,947 vs 7,154, respectively) but had far lower reporting odds ratios for 4 ADRs listed in Table 3: cough, dysphonia, pain, and dizziness. Interestingly, dizziness is listed in the drug product label for BUD/FORM under post-marketing experience as an ADR, but it is not mentioned as an ADR in the drug product label of FF/VI.12 Similarly, Table 4 shows that FP/SAL also yielded far lower reporting odds ratios for dyspnea, cough, pain, and dizziness than FF/VI. Dyspnea and dizziness are listed under post-marketing experience of FP/SAL; however, these adverse events are not addressed in the drug product label of FF/VI.13 These competitor findings suggest that patients taking FF/VI experience certain adverse events not commonly observed in its pre-approval clinical trials as they were not mentioned as potential adverse events in its drug label.
The primary goal of treatment for both asthma and COPD is to improve the patient’s quality of life because there is no definitive cure for either disease. To accomplish this, it is important to personalize medication regimens to maximize patient symptom relief while minimizing adverse events. Therefore, it is crucial to continually update medication safety profile as more data are available to be able to make an ideal treatment decision.
Limitations
Pharmacovigilance is an important component of evaluating and monitoring medication safety after initial drug approval. While there exists a wide variety of different databases that offer unique reports, FAERS is one of the most widely used and readily available to the public. Every database has their own pros and cons, and FAERS has the inherent limitation of being a voluntary self-reported dataset. Voluntary consumer reporting was abundant in our data, with 70% of our reports being from consumers and providing a different context compared to other studies. This information has the bias inherent to voluntary reporting data as patients who had negative experiences are more likely to actively report. However, as the medications of interest in this study are delivered via inhalers that are often used in out-patient settings, it is inevitable to see that a majority of reported ADRs are from the consumers themselves. Similarly, it is important to note that 87% of our reports are from North America (80% United States, 7% Canada) and 5% were indicated as “Country not specified.” Because the FAERS database captures reports submitted to the FDA, it is inevitable to have a majority of reports from North America.
Moreover, the general quality of the FAERS data is not best suited for a precise pharmacovigilance process. There is a possibility of duplicate reports and missing information, uncertainty of reporter identity, lack of causation relationship evidence, and accidental misinformation. We have attempted to counteract many of these limitations by removing duplicate reports and reports with missing ADR data. Finally, there is uncertainty that the reported ADRs are actually derived from the suspect medication (https://www.fda.gov/drugs/surveillance/questions-and-answers-fdas-adverse-event-reporting-system-faers, Accessed May 12, 2021). We present these data as signals for further investigation.
Due to these underlying limitations, it is not recommended to use FAERS data to calculate the actual incidence of adverse events. Therefore, a reporting odds ratio is the favored form of analysis to estimate reported associations as used in previous literature.22-24 One drawback of using reporting odds ratios is that a small number of reports may be more prone to be skewed by the paucity of information available, making comparison of 2 medications with significantly different numbers of reports difficult. To compensate with this potential flaw, we chose only medications with > 500 observations, as it has been previously concluded to be the minimum number of reports needed to perform disproportionality analysis with confidence for national databases.8
Lastly, our data may have a reporting bias affected by the Weber effect, which occurs when ADR reports peak at the end of second year after its approval, followed by a continuous decline thereafter.25 This is most relevant in our comparison of FF/VI and preexisting medications of the same class.
Conclusions
Post-marketing pharmacovigilance is needed to help all clinicians make appropriate clinical decisions. Pharmacovigilance provides safety insights that may have not been reported in clinical trials, and it can be used to better personalize patient therapy, optimize medication regimen, and ultimately improve patient health. Our analysis suggests that patients on UMEC or UMEC/VI may experience headache, pain, and signs of infection, as shown from clinical trials. FAERS data analysis on roflumilast also indicates the close resemblance of ADRs found from its clinical trials, both pre-approval and post-marketing trials. However, some disagreement was seen in the analysis of FF/VI. Dizziness and dyspnea were frequently reported as ADRs in FAERS for FF/VI, but these effects were not listed in its product label because they were not as frequently reported in clinical trials. Data mining with measures of disproportionality, as done in this study, provides good insight into the real-world frequency of different ADRs, thereby allowing the continuous update of medication safety profiles to optimize medication regimens.
Footnotes
- Correspondence: Sandra L Kane-Gill PharmD MSc, University of Pittsburgh, School of Pharmacy, PRESBY/SHY Pharmacy Administration Building, 3507 Victoria St, Mailcode PFG-01-01-01, Pittsburgh, PA 15213. E-mail: kane-gill{at}pitt.edu
Supplementary material related to this paper is available at http://www.rcjournal.com.
The authors have disclosed no conflicts of interest.
- Copyright © 2021 by Daedalus Enterprises




