

Abstract
Ventilator-induced lung injury (VILI) results from injury to the blood-gas barrier caused by mechanical ventilation. The determinants of VILI are more complex than originally thought, and include the nature, duration, and intensity of the exposure, as well the pattern of initial insult to the lung. Lung-protective mechanical ventilation founded on these basic principles resulted in improved hospital and long-term mortality. The purpose of this review is to provide a comprehensive assessment of the pathogenesis of VILI and its determinants. We also discuss the best preventive approach in patients with or at risk for ARDS and critically appraise the most recent evidence, expert opinion, and implementation of the acquired knowledge to the bedside.
Introduction
Marcello Malpighi first discovered the blood-gas barrier in 1661, when he noticed that the entire mass of the lung was an aggregation of membranes.1 Frank Low first demonstrated with electron microscopy, in 1952, that the blood-gas barrier consists of a single layer of alveolar epithelium and capillary endothelium, separated by a basement membrane.2 This delicate structure (Fig. 1) has an area approximately 1,000 square feet, a thickness of 0.2 μm, and can withstand transmural pressures up to 35 cm H2O.3 Mechanical injury to the blood-gas barrier is the hallmark of ventilator-induced lung injury (VILI). The key to understanding the pathophysiology of VILI lies in understanding the nature of the stress distribution in the lung and the cellular and molecular biology responses of the blood-gas barrier to tensile stress, among patients with and without already substantially injured lungs.

Electron microscopy of the blood-gas barrier. The arrow points to the extracellular membrane forming a cable around the blood alveoli. The arrowhead points to tissue separating adjacent capillaries. A: alveoli. B: Endothelium. C: Erythrocytes. (From reference 3, with permission.)
The Stress Distribution in the Lungs
The stress distribution in the lungs is more complex than initially thought. The interaction of the relatively rigid chest wall and abdomen with the lungs (Fig. 2A) is more influential in the topographic deformational stress and strain than the lung weight.4 Examination of laser confocal images of edematous, injured lungs indicates the presence of both collapsed and overdistended alveoli with a range of air pockets of differential radius surrounded by fluid and foam (see Fig. 2B). The presence of nonuniform gas pressures and/or surface tension may require high pressures to drive the foam out of the alveoli. Fluid-filled alveoli create areas of the lung with high surface tension.4 Understanding the pathophysiology of these flooded, unstable lung units and the airways that are repetitively opening and closing is central to the concepts of ventilator management and the prevention of VILI.5

A: Shape matching between an elastic cone (elastic solid, lung equivalent) and a rigid cylinder (chest wall equivalent) shows the nonuniform stress that is not determined by gravity in the lungs. B: Laser confocal images of subpleural alveoli of a normal lung (left) and a lung of a rat with ventilator-induced lung injury (right). (From reference 4, with permission.)
At the cellular level, the adaptation and responses of the alveolar cells are more complicated. From the mechanical standpoint, the normal blood-gas barrier, when stretched, first suffers disruption of pores in the endothelium, resulting in leakage of protein to the interstitial space. Upon higher stress insult or stress failure, both the endothelium and the alveolar epithelium become disrupted, with subsequent leakage of protein to the alveolar spaces, resulting in pulmonary edema.6
Determinants of VILI
The main determinants of VILI include the nature, duration, and the intensity of exposure, as well the first insult or hit to the lung (Table).7 The duration of exposure as an important element in VILI can be demonstrated from animal studies showing a clear relationship of the duration of injury (lung overdistention) and the overall microscopic structural abnormalities. In small animals, severe VILI can occur within 1 hour,7 whereas large animals may require longer duration (up to 24 h).
Determinants of Ventilator-Induced Lung Injury7
Volutrauma
The intensity of exposure is another key determinant of VILI. The principal role of membrane strain, or volutrauma, in rats with or without strapped chests (limiting their respective thoracoabdominal excursions) is elegantly described by Dreyfuss et al.8 This study demonstrated that rats with low tidal volumes (VT) developed no pulmonary edema, despite high airway pressures. However, when the investigators used negative pressures (emulating the iron lung), high VT was still the main determinant of pulmonary edema.
Functional Residual Capacity and the Size of the Available Lung
A particularly intriguing concept is the concept of the “baby lung,” first introduced by Gattinoni.9 Baby lung is the small portion of aerated lung with near-normal compliance, which is open during the end-expiration phase (see Fig. 2B). This concept emphasizes the novel idea that the ARDS lungs are not stiff, but small.9 Therefore, the size of the baby lung could render the injured lung more susceptible to volutrauma, introducing the concept of relativity in VILI based on the size of the baby lung.
Atelectrauma and Heterogeneity
Heterogeneous lung areas with atelectasis, consolidation, and edema aggravate the development of VILI. Repeated opening and closing of surfactant-deficient lung units renders adjacent zones susceptible to shear stresses, creating a vicious cycle of VILI, both at high and low VT (“atelectrauma”).7 The epiphenomenon of liquid and foam accumulation further aggravates the VILI.10
End-Inspiratory Lung Volume
As shown in Figure 3, selecting a low end-inspiratory volume (near the relaxation volume of the lung), promotes derecruitment and possibly further lung damage, whereas selecting a high end-inspiratory lung volume toward the total lung capacity increases the risk of overdistention injury.10

A graph with the therapeutic end points in an injured lung. The 2 extremes represent a volume near the total lung capacity on the right and near the relaxation volume on the left. (From reference 10, with permission.)
PEEP
The effect of higher PEEP was shown to decrease the amount of edema in multiple animal studies. This may be multifactorial, given the fact that higher PEEP may lead to the use of lower VT and capillary filtration (secondary to reduced cardiac output), as well as preservation of the surfactant production, and does not prevent changes in the permeability of the endothelium.7 Both the volume history (recruitment) and chest wall compliance (see above) strongly influence the required PEEP setting necessary to prevent atelectrauma, complicating clinical management and interpretation of clinical research studies (see below).
Inspiratory Flow and Flow Profile (Strain Rate)
The inspiratory flow is another variable that one has to keep in mind when adjusting ventilator settings. Rich et al11 have demonstrated dramatic impact of high inspiratory flow on healthy animal lungs. It appeared that the higher the inspiratory flow for given pressures, the higher was the resultant VILI.11 Furthermore, a study in small animals demonstrated increased microvascular injury with higher flows.
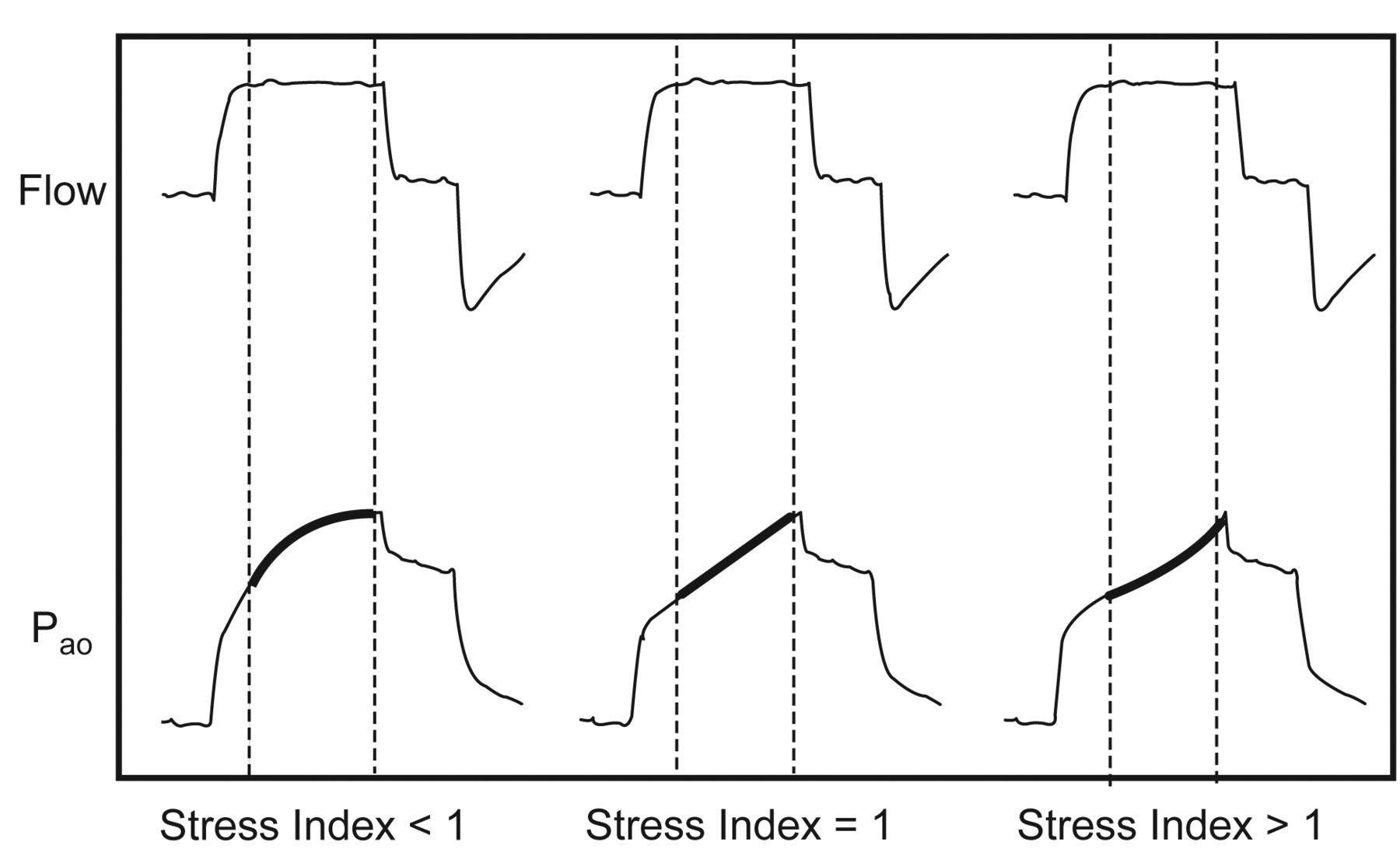
Another important concept is that having a constant flow permits the estimation of the stress index (Fig. 4). The rate of change in airway opening pressure, reflected on the stress index, correlates with the rate of change of lung elastance (or compliance); however, there is limited evidence that a stress-index-guided PEEP selection strategy (titrating PEEP with a goal stress index of around one) may reduce the risk of alveolar hyperinflation and subsequent VILI.12

Ventilator pressure and flow curves indicating the correlation with the stress index. (From reference 12, with permission.)
Breathing Frequency
The selection of breathing frequency is a fundamental concept of mechanical ventilation (MV). Animal studies have shown that for the same VT/strain, higher breathing frequencies intensify VILI.13 Also, animal studies have demonstrated that lungs ventilated at low breathing frequencies and high peak pulmonary artery pressures produced less edema and less perivascular hemorrhage than did those ventilated at higher breathing frequencies and identical peak pulmonary artery pressures, with mean airway pressure and VT matched between groups.14 However, in clinical practice, ventilator rate often needs to match the patient's demand. In most ventilators, the inspiratory to expiratory time ratio is dependent on the rate, given that the inspiratory time is fixed. This makes the expiratory time variable and susceptible to the patient's demand rate. In continuous mandatory ventilation modes of MV, in which inspiratory time is fixed, a patient-triggered rate far in excess of the set rate increases the inspiratory/expiratory ratio, inadvertently leading to functional inverse ratio ventilation, dynamic gas-trapping, and excessive end-inspiratory lung volume. Expert opinion suggests that most patients with respiratory failure will require a rate between 20 and 30 breaths/min according to their metabolic needs.10
Vascular Pressures
The role of vascular pressures as a determinant of VILI is a concept based on the interdependence of the vessels with the lung parenchyma.7 At higher pressures, MV expands the physiologic West lung zone one (alveolar pressure > arterial > venous), promoting higher hydrostatic pressures and pulmonary edema.7 Furthermore, animal data suggest that there is an interplay with hemodynamic and airway pressure profiles, with higher changes in pulmonary artery pressures during MV being associated with increased vascular permeability, greater degrees of pulmonary hemorrhage, and edema.15
Initial (“First Hit”) Injury
In accordance with the multiple-hit theory of ARDS development, inflammation induced by sepsis, tissue injury, and transfusions/fluid overload renders the lungs susceptible to the “second hit” of MV and development of VILI.16 In particular, preexisting lung alterations (edema, atelectasis, pneumonia) make the diseased lung much more susceptible to mechanical injury.7
Lung-Protective Ventilation in Patients With ARDS
Prevention of VILI with lung-protective low VT MV is the cornerstone of ARDS management. Six randomized clinical trials addressed the lung-protective MV strategy,17–22 3 of which showed mortality benefit.17–19
Differences in methodology among the clinical trials might have contributed to different results. In 2 positive trials, the Brazilian single-center study,17 and the ARDS Network trial,18 there was a larger difference in the VT settings between the treatment groups (conventional treatment VT of approximately 12 mL/kg vs low VT arm 6 mL/kg), compared to the 3 negative trials (traditional treatment VT of approximately 10 mL/kg vs low VT arm 7 mL/kg).20–22
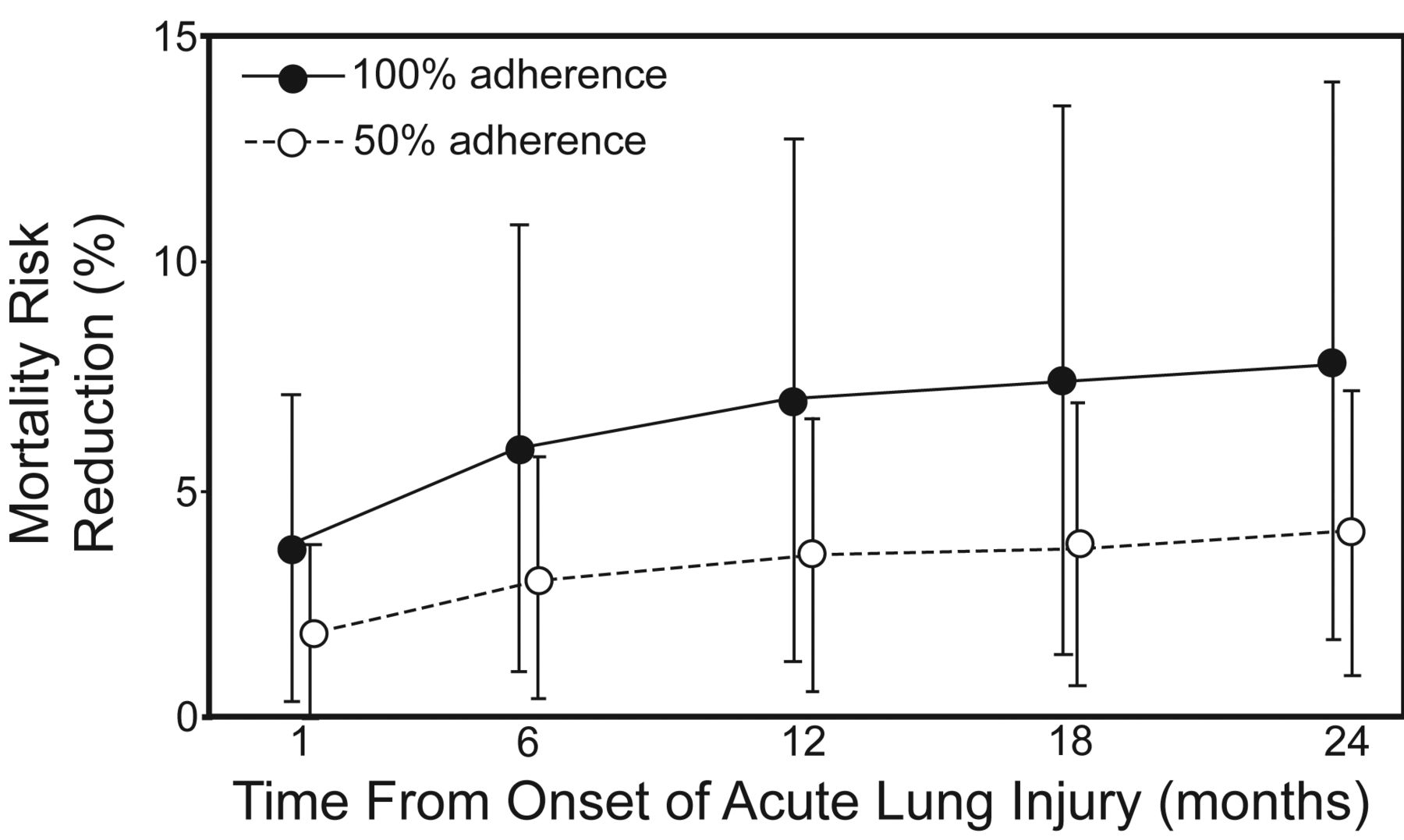
Not only has in-hospital survival been shown to be higher using lung-protective strategy, but also long-term survival rates. In a prospective cohort of 485 mechanically ventilated ARDS patients, the 2-year survival was greater in patients whose adherence to volume-limited and pressure-limited MV was greater than 50%, compared to those with no adherence to the lung-protective MV strategy (Fig. 5).23
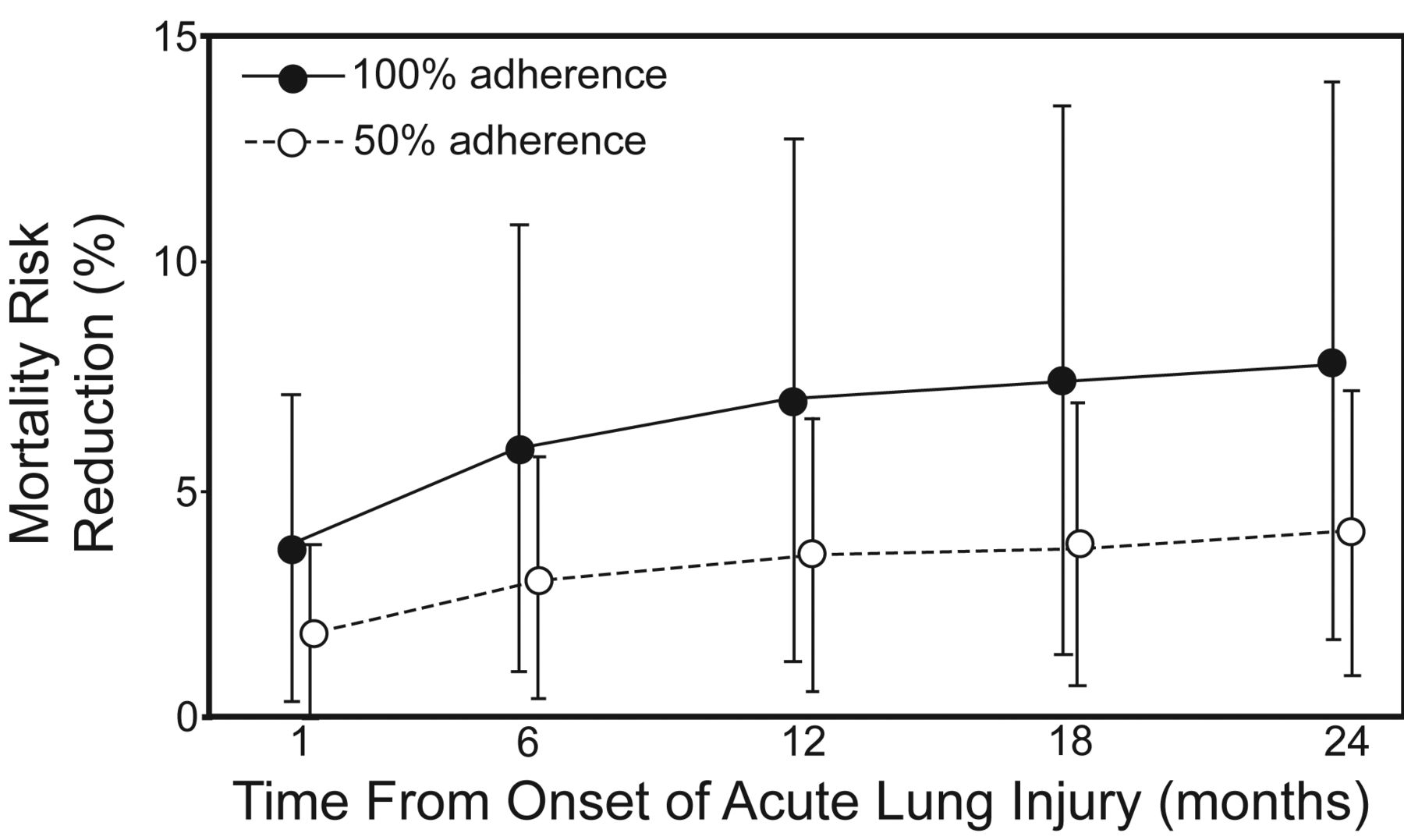
Predicted absolute risk reduction in mortality by adherence to lung-protective ventilation. (From reference 23, with permission.)
Patient-ventilator asynchrony often limits the use of low VT in patients who most need it (ie, patients with ARDS with high minute ventilation requirements due to metabolic acidosis and high dead space). Adjustment of ventilator settings and sedative agents are only modestly effective in limiting patient ventilator asynchrony, often necessitating the use of neuromuscular blockade. A recent multicenter randomized trial addressed the use of neuromuscular blockers in patients with ARDS.24 Patients who received cisatracurium besylate for 48 hours had a reduction in the 90-day mortality, compared to those who received placebo, after adjusting for baseline PaO2/FIO2, plateau pressure, and the Simplified Acute Physiology Score (SAPS II). It also increased the time off the ventilator, and, importantly, did not increase muscle weakness.
The 6 randomized clinical trials mentioned above have not provided much information regarding the best PEEP to use in patients with ARDS. Recently, a meta-analysis of 3 other randomized clinical trials with a total of 2,299 patients showed that there was no reduction in mortality in the group of patients who received higher PEEP levels.25 However, in a subgroup analysis of this meta-analysis, higher PEEP was associated with potential benefit in patients with more severe hypoxemia (PaO2/FIO2 < 200 mm Hg) and potential harm in those with less severe hypoxemia (PaO2/FIO2 200–300 mm Hg). As discussed above, individual PEEP setting taking into consideration lung recruitment and chest wall compliance, with the assessment of stress index and in some cases pleural pressure measurement by esophageal balloon, may prove to be more effective than any set PEEP strategy.26 Many experts recommend a recruitment maneuver (slow lung inflation to near total lung capacity, a sigh) to optimize stress distribution within the lung prior to setting optimal PEEP strategy.27,28
Lung-Protective Ventilation in Patients at Risk for ARDS
The evolution of critical illness follows a nonlinear pattern typical of other complex systems3 where small changes (positive or negative) that occur before “phase transition” can have a profound effect on critical illness trajectory (for example, timing of appropriate antibiotics, meticulous fluid resuscitation during early shock, thrombolytic therapy or angioplasty for myocardial infarction, and prevention of gastric aspiration during endotracheal intubation). The implementation of preventive and therapeutic strategies early in the course of critical illness is crucial to patient outcome.4 A similar “golden hours” paradigm can be used for early treatment and prevention of VILI (before the development of full blown ARDS).
Observational Studies in the ICU
The first observational study addressing ventilator-associated lung injury in patients without ARDS was done at Mayo Clinic.29 Ventilator settings were highly variable, with many of the 332 patients without ARDS at the time of intubation being exposed to very high VT (> 12 mL/kg of predicted body weight [PBW]). The risk factors associated with the development of ARDS after initiation of MV were the use of large VT in a dose-response manner (odds ratio 1.3 for each mL above 6 mL/kg of PBW, P < .001) (Fig. 6), as well transfusion of blood products, acidemia, and a history of restrictive lung disease.
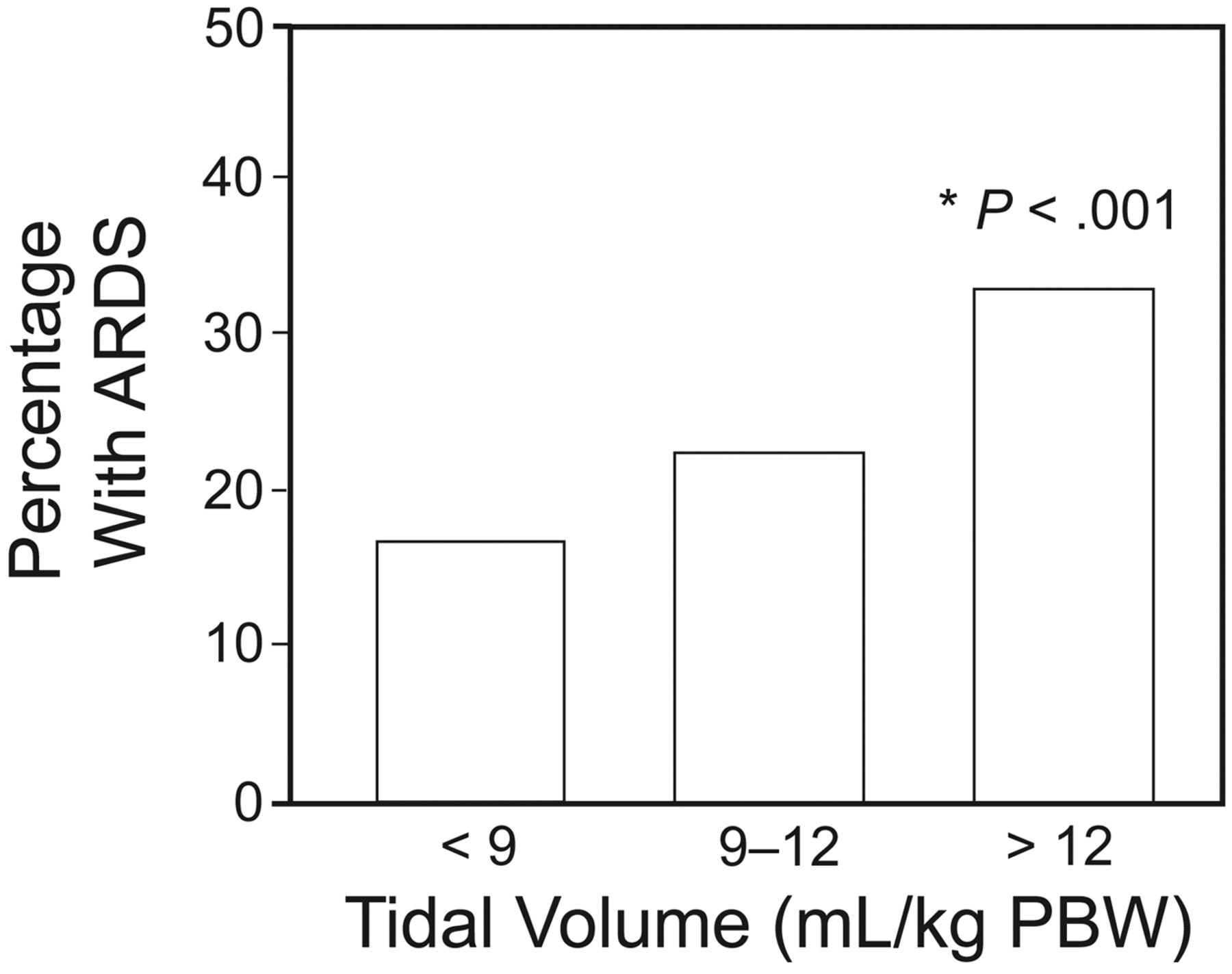
Percentage with ARDS versus tidal volume (VT). PBW = predicted body weight. * Adjusted P value from a multiple logistic regression model. (From reference 29, with permission.)
In the following year, a multicenter study with 3,261 mechanically ventilated patients also showed the association of large VT (odds ratio 2.6 for VT > 700 mL) and high peak airway pressure (odds ratio 1.6 for peak airway pressure > 30 cm H2O) with the development of ARDS after initiation of MV, after adjusting for baseline characteristics and underlying ARDS risk factors.30 Most recently, Lellouche et al reported on the harmful effect of high and traditional VT ventilation in post cardiac surgery patients.31
These studies highlighted the practice of setting VT according to actual, rather than PBW as an important barrier to lung-protective MV; exposing women, as well as shorter and obese patients, to higher risk. However, height and sex are better predictors of lung size than is actual body weight; therefore, PBW should be used to calculate VT for mechanically ventilated patients, in order to avoid VILI.10 A pragmatic approach to initial lung-protective VT settings is 450 mL for men and 350 mL for women, with corresponding increase in breathing frequency to match minute volume requirements (up to 30–35 breaths/min in patients with shock or severe acidosis).32
Randomized Controlled Trials in the ICU
In 1990, Lee et al33 randomized intubated surgical ICU patients to receive either VT of 6 mL/kg or 12 mL/kg. Patients who received the lower VT had a reduction in the duration of MV and surgical ICU stay. Twenty years later, Determann et al34 performed a randomized controlled trial comparing MV using conventional VT (10 mL/kg) versus lower VT (6 mL/kg). The study was stopped prematurely for safety reasons, since the development of ARDS was significantly higher in the conventional VT group (13.5% vs 2.6%, P = .01) (Fig. 7).
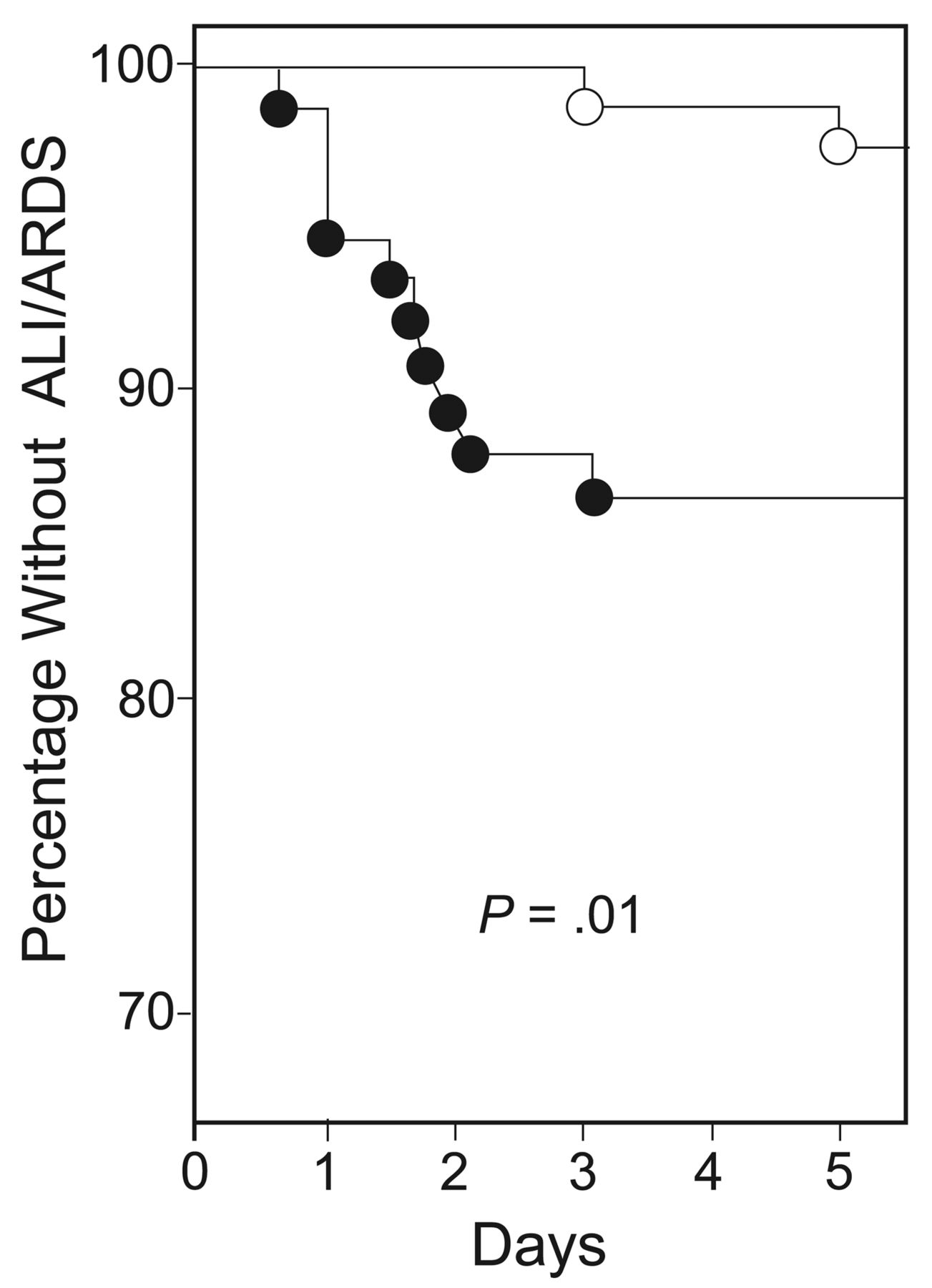
Kaplan-Meier curve of incidence of acute lung injury of patients ventilated with conventional tidal volume (VT) (solid circles) versus lower VT (open circles). (From reference 34, with permission.)
Observational Studies in the Operating Room
Studies involving lower VT were being done not only in the ICU setting, but also in the operating room. In 2006 Fernandez-Perez et al35 analyzed a cohort of patients undergoing pneumonectomy, and found an association between larger intraoperative VT (odds ratio 1.56 for each mL/kg increase) and a higher risk of post pneumonectomy respiratory failure, with ARDS being the most common reason. In 2009 the same institution performed a prospective, nested case-control study with 4,420 patients without ARDS undergoing high-risk elective surgeries. The study showed an association between the mean intraoperative peak airway pressure and the development of an acute lung injury, but not between (already modest) VT and acute lung injury.36
Randomized Controlled Trials in the Operating Room
Several randomized studies have been done in the operating room and during postoperative states, addressing low VT and the risk for development of ARDS, with either no effect or modest benefit of lung-protective MV. Importantly, low VT ventilation was consistently safe.37
Summarizing the above studies, Shultz et al37 have proposed to ventilate patients with normal lungs and no ARDS risk factors with VT < 10 mL/kg of PBW, plateau pressure < 15–20 cm H2O, and PEEP ≥ 5 cm H2O; and patients with compromised lungs (interstitial lung disease, lung resection, severe pneumonia, and edema) and/or the presence of ARDS risk factors (sepsis, aspiration, transfusions) with VT of 6 mL/kg PBW, plateau pressure < 15–20 cm H2O, and PEEP ≥ 5 cm H2O.
Recently, Yang et al38 randomized 100 patients undergoing one-lung ventilation during elective lobectomy to either the conventional ventilation group (FIO2 1.00, VT 10 mL/kg, PEEP = 0 cm H2O, and volume-controlled ventilation) or the protective ventilation group (FIO2 0.50, VT 6 mL/kg, PEEP = 5 cm H2O, and pressure-controlled ventilation). The incidences of PaO2/FIO2 < 300 mm Hg, lung infiltration, or atelectasis were significantly lower in the protective strategy (4% vs 22%, P < .05).
A very recent meta-analysis published in JAMA of observational and randomized studies of mechanically ventilated patients without ARDS strongly suggests that the benefit of lung-protective MV extends to all mechanically ventilated patients, with potential for prevention of lung injury of patients who do not already have it.39
Putting It All Together
After having all this evidence favoring the practice of low VT MV, the question that arises is, why did we formerly use high VT in the first place? Impairment of oxygenation is commonly seen from atelectasis during anesthesia and MV. Low alveolar ventilation-perfusion and true shunt are the major functional causes for this; these are thought to be due to rapid collapse of alveoli during induction of anesthesia, as well as extensive airway closure. Possible contributing mechanisms for alveoli collapse and airway closure are loss of respiratory muscle tone and gas reabsorption.40
Furthermore, it was hypothesized that VT could be a significant determinant in the increased alveolar-arterial oxygen difference during MV. Therefore, in 1973 Visick et al41 proposed using large VT in order to increase PaO2. However, the effectiveness of this approach has not been confirmed by others.42,43 In fact, what has been shown to prevent or reduce atelectasis is the avoidance of high FIO2 during both induction and maintenance of anesthesia.44,45 Equally or more important, “vital capacity” maneuvers or “recruitment” maneuvers have been shown to recruit atelectatic lung regions.27,46,47 A randomized controlled trial28 tested the same hypothesis of alveolar recruitment maneuver on obese subjects undergoing laparoscopic bariatric surgery. Patients in the recruitment group (received up to 4 sustained lung inflations with peak inspiratory pressure up to 50 cm of H2O, followed by ventilation with PEEP of 12 cm of H2O) had a significantly higher intraoperative PaO2/FIO2, compared to the control group (ventilated in a standard fashion with PEEP of 4 cm of H2O) (P < .01).
Applying the acquired knowledge to the bedside using quality improvement interventions is extremely important in the aim to prevent VILI. Yilmaz et al48 implemented a quality improvement intervention in mechanically ventilated patients without preexisting ARDS that consisted of a Web-based teaching tool and a respiratory therapy protocol limiting the initial VT according to PBW in all patients (using a simple nomogram available on all ventilators). After the intervention, a large decrease in VT and in peak airway pressure, as well as in the frequency of new ARDS cases and mortality, was observed
Summary
In summary, normal (low) VT ventilation is safe and effective in maintaining gas exchange in patients without ARDS. In mechanically ventilated patients at risk of ARDS, exposure to high VT increases the frequency of ARDS. In patients with established ARDS, exposure to high VT increases mortality rates. Height and sex are better predictors of lung size than actual body weight; thus, PBW should be used to calculate the appropriate VT in mechanically ventilated patients. New strategies based on direct measurement of the size of the baby lung (functional residual capacity) may further optimize VT in individual patients.49 Neuromuscular blockade may be necessary to prevent ventilator asynchrony and facilitate lung-protective MV in patients with high metabolic demand or increased dead space. While optimal PEEP management remains controversial, the authors recommend a recruitment maneuver followed by minimum PEEP setting to prevent both overdistention (plateau pressure > 30, stress index > 1) and derecruitment (stress index < 1). Ongoing studies will further delineate the role of esophageal manometry in guiding optimal PEEP setting, particularly in patients with presumed high chest wall compliance (obesity, abdominal distention).
Footnotes
- Correspondence: Ognjen Gajic MD, Division of Pulmonary and Critical Care, Mayo Clinic, 200 First Street SW, Rochester MN 55905. E-mail: gajic.ognjen{at}mayo.edu.
Dr Gajic presented a version of this paper at the 51st Respiratory Care Journal Conference, “Adult Mechanical Ventilation in Acute Care: Issues and Controversies,” held September 7 and 8, 2012, in St Petersburg, Florida.
The authors have disclosed no conflicts of interest.
- Copyright © 2013 by Daedalus Enterprises
References
Discussion
MacIntyre:
Your list of VILI mechanisms did not include flow, but faster flow is more dangerous than slower flow.1 Could you comment on that?
Gajic:
The question about flow is how fast does the strain occur; that is, the delta in time of strain. I'm not aware of any clinical studies, but animal data suggest that lower flow may decrease VILI.1 To extrapolate to clinical medicine, the square wave profile might be preferable but might be less well tolerated than a decelerating flow profile, unless the patient is paralyzed. What do you think?
MacIntyre:
The intensity of exposure could be important; I don't know.
Gajic:
Maybe in theory it should work having a constant flow with a slower rate of strain with each breath, which, in theory, should cause less injury. However, the way to do it, which is changing the square wave profile, is just intolerable for the patient, so we can't do it. That's a patient-ventilator synchrony question.
Marini:
Experimentally, in small and large animals, flow does make an important difference in a nonhomogeneous lung. And not only flow, but the rate of development of inspiratory shear stresses: the flow profile. No one has looked at it in the clinical setting, as far as I know, and it's obviously a very difficult thing to look at. The peak flow is conditioned by whether you're using pressure control or flow control, and pressure control is the most damaging in small animals. This should make us think. Neil asked a good question.
Kallet:
One of the things that gets confused about this with pressure control and lung-protective ventilation is that, as you turn the pressure control level down to keep VT at 6 mL/kg, it drops your peak inspiratory flow, even when the patient is triggering the ventilator. We studied this at San Francisco General Hospital many years ago,2 and the peak flow was about 100 L/min with VT of 10 mL/kg. But when we did this with the ARDS Network3, the peak flow was 50 L/min and the patients sometimes were more asynchronous on pressure control as we turned the peak pressure down to 20 cm H2O. It's an important point. A separate point is whether someone with a high respiratory drive who wants to be hyperpneic will tolerate ventilator settings that limit their flow. That gets us into other problems with sedation, so we have to be careful.
MacIntyre:
With our protocols at Duke University I have convinced the medical and surgical ICUs and the critical care unit that smaller VT is a good idea, but I still run into a stone wall with the thoracic surgery unit, where the protocol they submitted to me says that post-operative thoracotomy patients should receive VT of 10-12 mL/kg. Are there any data that support that?
Gajic:
Not really. Lellouche, in Canada, conducted an observational study,4 but it's quite well adjusted. They found that in cardiovascular surgery patients—not lung resection patients—even in those patients the exposure to these outrageous 10+ mL/kg tidal volumes is associated with worse lung function and poor outcomes. They better change their practice.
MacIntyre:
I agree. I wanted to be sure they have no data supporting that practice.
Gajic:
No. A recent systematic review5 summarized the randomized and observational studies in patients without ARDS, many of which were in cardiovascular surgery patients, and most of the studies suggested some benefit from lung-protective mechanical ventilation in cardiac surgery patients.
Schmidt:
Why do we use a different VT when ventilating patients with normal lungs? I breathe right now at about 5-7 mL/kg, but why do we make it so difficult for the practitioners? To receive information about ARDS risk and what changed this risk in the operating room will sometimes take me a couple of hours. Why not ventilate everybody in the operating room with 6 mL/kg and PEEP of 5-10 cm H2O? This would make it easier for every practitioner and would minimize injurious ventilation.
Gajic:
That's what we've done. The requirement that ARDS is present before adopting low-VT ventilationmakes it extremely difficult, because of who will agree what ARDS is. So that's been overcome in many places; all ICU patients are ventilated with 4-8 mL/kg predicted body weight, which is basically the ARDS Network protocol, regardless of the presence or absence of ARDS, and that makes it much easier. In the operating room we have moved in that direction; the average mean VT in the Mayo Clinic operating room is now around 7 mL/kg predicted body weight. There's no reason to use high VT, since PEEP can overcome derecruitment that occurs with paralysis during surgery.
Branson:
I was intrigued how quickly you go to neuromuscular blockade in your hypoxemia algorithm. Every July the new critical care fellows ask me to give a lecture about the things I think they should understand about mechanical ventilation. And every year the medical ICU—where I don't work—won't allow the respiratory therapists to use adaptive pressure ventilation, and every year within a week the pharmacists come to me and say “You have to do something; we're running out of propofol.” Rich, do these patients tolerate lower VT without additional sedation? I know what the literature says, but that just isn't our practical experience.
Kallet:
In my experience, I don't think the majority of critically ill patients can tolerate a physiologic VT without some pharmacologic support. That being said, I have come across some who do very well on 6 mL/kg without it.
One of the problems I think we've had in these conversations is that we always think about the critically ill patient who is very well sedated. We tend to forget that breathing is not just metabolics; it's very heavily intertwined with the expression of emotion. Being of the Hebrew persuasion, I can say modestly that I have this covered in spades! Normally we breathe at 6 or 7 mL/kg. But we also frequently sigh; and fear, pain, et cetera all impact our VT demand.
We also tend to forget that the perception of dyspnea is directly related to the amount of tension in the respiratory muscles relative to the corresponding displacement of the chest, and when that's restricted in some way, it feeds back to the brain as dyspnea and causes an automatic response to increase respiratory drive. In a critically ill and in pain person, if all of a sudden their breathing gets worse—these things can change on a minute-by-minute basis—I think it's very hard clinically to say that anyone's going to breathe at 6 mL/kg under those conditions, without sedation or analgesia. And sometimes, as a compromise, it becomes a matter of harm reduction.
At our hospital we start everyone at approximately 8 mL/kg, we're pretty good at keeping to 6 mL/kg for ARDS, with good mortality outcomes. However, I haven't particularly tracked how much propofol we consume to achieve that, which is Rich's point. I think one interpretation of our research on work of breathing with lung-protective ventilation3 might suggest that with pressure ventilation you might possibly get away with less sedation because the patient can achieve a VT that's more satisfying with moderately more effort, but the consequence is somewhat less lung-protective ventilation, which is probably not helpful. Also I don't think patients need to be perfectly synchronous. There has to be some type of balance between comfort, lung protection, and not pickling their brains with sedation.
Hess:
Rich, I know you're going to talk about asynchrony later this morning, but one of the things that has struck me over the years—Taylor Thompson and I wrote an editorial about this a few years ago in Critical Care Medicine6—is that I think a lot of asynchrony is unrelated to the ventilator and how we set the ventilator. It's because the patient is acidotic, the patient is in pain, the patient is anxious. To address some of those issues we need to use adequate pain control, we may need to use anxiolytics, and we need to correct the acidosis, and we miss some of those things because we are trying to somehow adjust the ventilator so that it can address the problem. I would go back to the Papazian study,7 which has certainly been informative to me in my practice; there may be a role for paralysis in the first 48 hours after the patient is intubated.
Gajic:
But 48 hours of infusion may be too long and is rarely needed, in my opinion. I've talked with them as well, and for a study the problem is how you read it, how you apply evidence to your actual practice, and how you develop your protocol. To maximize the chance of benefit in a clinical trial the researchers need to have a large difference in exposure between the 2 study groups.
Rather than a prolonged continuous infusion I often use single bolus doses of NMB [neuromuscular blocker], to eliminate ventilator asynchrony and to facilitate recruitment maneuvers and assess lung mechanics. In patients with severe hypoxemia I use a single dose of NMB right after intubation, to assess lung mechanics and find out what's going on. Sometimes I find evidence of auto-PEEP and COPD and other things I didn't know about. Chest wall compliance can be more easily appreciated. This is both a therapeutic and a diagnostic maneuver, and I find that as the patient emerges from that single dose of NMB, they start synchronizing with the ventilator better. But I use only one or two bolus doses, rarely more, and it is exceptionally rare to use a continuous infusion of NMB.
Hess:
One of the things that struck me in the Papazian paper,7 was that, as I recall, the group of patients who were not paralyzed had a higher incidence of pneumothorax, which suggests that there was injury from the patient tugging hard to breathe against the ventilator.
Gajic:
I think that's the best explanation. It's very difficult to come up with a mechanism other than mechanical injury to explain the benefit of neuromuscular blockade. That's the most likely.
Branson:
I'd like to ask you about your paper8 in Respiratory Care on FIO2. How important is FIO2 to lung injury? We often see trauma patients on FIO2 of 0.40 to 0.45 and with a PaO2 of 180 mm Hg. Is it just the O2 in the lung that's toxic, or can hyperoxemia cause problems in distal organs?
Gajic:
That's an excellent question about O2 toxicity. I think there's evidence of both. There is direct toxicity to the lungs: prolonged exposure causes diffuse alveolar damage and acute lung injury, but the potential harm of hyperoxia is probably not limited to the lung. In the first few hours of acute shock O2 is beneficial. As soon as reperfusion injury sets in, getting O2 to the fire can make the condition worse. This is analogous to the effect of thrombolytic therapy or angioplasty in the setting of acute MI [myocardial infarction] or stroke, which lessens dramatically after the first 3-6 hours from the onset.
Schmidt:
Is there clinically important O2 toxicity in adults? It's described in every textbook, but the supporting evidence is poor or nonexistent. It might be something we're always looking for and have never found. In the operating room, 100% O2 has been shown to be better for wound healing. Why do we often insist on decreasing the FIO2 to 0.50 in ventilated patients?
Gajic:
Bronchoscopic studies9 show that even short-term exposure to an FIO2 of 0.50 causes distinct pathophysiologic changes in human lungs. This is subclinical, obviously. How important it is in the context of critical illness is unknown. High FIO2 is probably beneficial for a short time early in the course of acute ischemia, so short-term use of high intraoperative FIO2 to enhance wound healing is probably fine. On the other hand, high FIO2 promotes atelectasis and may not be beneficial in some circumstances. I don't know that the data are very clear that you would always want to ventilate somebody in the operating room with an FIO2 of 1.0.
Marini:
A paper from England in Critical Care Medicine reviewed the data,10 and they were quite convinced that down-regulating FIO2 is very important. If so, accommodating patients to lower levels of oxygen saturation may be of immense benefit. I wrote the editorial11 for that paper, and I found the reviewed data very interesting. It's quite suggestive—not a confirmation by any means, but there is intriguing evidence that FIO2 is a co-factor that combines with purely mechanical stresses to cause problems.
Gajic:
I would look at it like high VT and low VT: why use outrageously high O2 unless you have a specific reason? If you have a specific reason, then for a short time that's great, but otherwise I don't see the reason to use potentially toxic high FIO2 to keep a high O2 saturation.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Introduction
- The Stress Distribution in the Lungs
- Determinants of VILI
- Lung-Protective Ventilation in Patients With ARDS
- Lung-Protective Ventilation in Patients at Risk for ARDS
- Putting It All Together
- Summary
- Footnotes
- References
- MacIntyre:
- Gajic:
- MacIntyre:
- Gajic:
- Marini:
- Kallet:
- MacIntyre:
- Gajic:
- MacIntyre:
- Gajic:
- Schmidt:
- Gajic:
- Branson:
- Kallet:
- Hess:
- Gajic:
- Hess:
- Gajic:
- Branson:
- Gajic:
- Schmidt:
- Gajic:
- Marini:
- Gajic:
- References
- Figures & Data
- Info & Metrics
- References