

Abstract
Diffuse cystic lung diseases (DCLDs) are a group of diverse pulmonary disorders with varying pathophysiology that are characterized by the presence of thin-walled, air-filled spaces within lung parenchyma. High-resolution computed tomography plays a crucial role in the evaluation of DCLDs, and cyst characteristics such as morphology, distribution, and the presence of other associated radiologic findings can help distinguish between different DCLDs. DCLDs can be classified according to their underlying pathophysiology as neoplastic, genetic, lymphoproliferative, infectious, associated with other forms of interstitial lung disease, or related to smoking. In this review we will provide a clinical overview on the most common DCLDs that are encountered in clinical practice: lymphangioleiomyomatosis, pulmonary Langerhans cell histiocytosis, Birt-Hogg-Dubé syndrome, and lymphoid interstitial pneumonia/follicular bronchiolitis, with a focus on practical aspects that can help clinicians in the optimal diagnosis and management of patients with DCLDs.
- lymphangioleiomyomatosis
- pulmonary Langerhans cell histiocytosis
- Birt-Hogg-Dubé syndrome
- lymphoid interstitial pneumonia
- follicular bronchiolitis
Introduction
Diffuse cystic lung diseases (DCLDs) are a heterogeneous group of pulmonary disorders that are characterized by multiple air-filled spaces, or cysts, within the lung parenchyma.1 Cysts are thin-walled (< 2 mm wall thickness), spherical, air-filled lucencies interfaced with normal lung tissue (Table 1).2 Critical review of cyst characteristics such as shape, size, wall thickness, and distribution on high-resolution computed tomography (HRCT) plays a major role in the evaluation of DCLDs.
Fleischner Society Definitions of Air-Space Lucencies on CT Scan
The exact mechanisms of cyst formation in DCLDs are not well elucidated and likely vary depending upon the underlying disease. Broadly, there are 3 major processes that have been linked to the development of cysts: (1) dilation of air spaces as a result of one-way obstruction in small airways leading to air entering but not exiting air spaces,3 (2) ischemia causing necrosis of small bronchioles,4 and (3) remodeling from matrix-degrading proteolytic enzymes.5 We have previously proposed a pathophysiology-based classification of DCLDs (Table 2). In this review, we will focus on the major DCLDs that a clinician is most likely to encounter in practice: lymphangioleiomyomatosis (LAM), pulmonary Langerhans cell histiocytosis (PLCH), Birt-Hogg-Dubé syndrome (BHD), and lymphoid interstitial pneumonia (LIP)/follicular bronchiolitis (FB).
Classification of DCLDs
Lymphangioleiomyomatosis
Lymphangioleiomyomatosis (LAM) is a rare DCLD that predominantly affects women.6 The average age at diagnosis is ∼ 35 y,1 but it has been reported in all age groups ranging from teenagers to elderly females.7,8 LAM occurs in 2 forms: in patients with the inheritable disease tuberous sclerosis complex (TSC-LAM), and in a sporadic form in patients without TSC.9 The estimated prevalence of LAM is 5–8 per million women,10 although that is almost certainly an underestimate.
Pathophysiology
The central event driving the pathogenesis of LAM is a mutation in the TSC genes. Both TSC1 and TSC2 mutations have been described in patients with TSC-LAM, whereas only TSC2 mutations have been described in patients with sporadic LAM. TSC mutations are present in the germline in cases of TSC-LAM, while sporadic LAM patients have somatic mutations that is mutations limited to the abnormal cells only. This explains the transmission of TSC-LAM from one generation to the next, which is not seen in sporadic LAM.11 TSC1 and TSC2 translate into large proteins called hamartin and tuberin, respectively, which negatively regulate the intracellular serine/threonine kinase mTOR signaling pathway, which is responsible for regulating cell size, proliferation, and survival by assimilating signals from growth factors, energy, and stress.12 Therefore, hamartin or tuberin deficiency or dysfunction causes increased activity of mTOR, leading to inappropriate cell growth and proliferation.13 Activated mTOR pathway drives proliferation of abnormal smooth muscle cells (LAM cells), arising from an unknown source of origin, and abnormal angiogenesis and lymphangiogenesis (in part) through expression of vascular endothelial growth factors (VEGF-C and VEGF-D),14 which leads to migration of LAM cells through the blood and lymphatic channels and ultimate deposition in the pulmonary parenchyma. In this regard, LAM behaves as a low-grade destructive metastatic neoplasm.15 Once in the pulmonary parenchyma, the LAM cells lead to the formation of lymphatic clefts through a disordered remodeling process that occurs in response to inappropriate lymphangiogenic signaling with secretions of matrix-degrading enzymes, thus leading to the progressive cystic destruction of the lung tissue.16–18 Although the exact origin of LAM cells remains unknown, leading candidates include the uterus,19 kidneys,14 and pelvic lymph nodes. Given the striking female predominance of LAM, the role of female sex hormones such as estrogen in the pathogenesis of LAM has been proposed; estrogen has been shown to accentuate the metastatic potential of LAM cells in preclinical cell culture models and animal studies.20,21
Clinical Presentation
Patients with LAM typically have one of the following modes of presentation: gradually worsening dyspnea, spontaneous pneumothorax, or incidental discovery of cysts on imaging performed for unrelated reasons. Pneumothoraces are seen in about 60–70% of LAM patients and tend to be recurrent.22 The most common pulmonary function test (PFT) abnormality in LAM is air-flow obstruction followed by decreased diffusion capacity of the lung for carbon monoxide (DLCO).23,24 It has been suggested that patients who present with dyspnea may have more advanced disease and a greater risk of mortality as compared to patients presenting with a pneumothorax.25,26 This likely represents a form of lead-time bias as patients presenting with a pneumothorax tend to get diagnosed at a younger age compared to patients presenting with dyspnea.27
Radiology
Chest computed tomography (CT) scans in LAM classically show the presence of multiple, well-defined, round, thin-walled cysts, usually 2 mm to 2 cm in size, scattered throughout both lungs (Fig. 1, Table 3).28–30 Other less common findings include multifocal micronodular pneumocyte hyperplasia, a hamartomatous process of the lung that manifests radiologically as multiple small, discrete, solid and ground glass nodules and is seen mainly in patients with TSC-LAM.31 In patients with lymphatic involvement, interstitial chylous infiltrates and chylous pleural effusions might be present. Other extrathoracic manifestations of LAM include fatty tumors called angiomyolipomas (AMLs), which are seen most commonly in the kidneys, lymphadenopathy, fluid-filled cystic structures in the axial lymphatics (lymphangioleiomyomas), and chylous ascites.32–35 Renal AMLs can be seen in approximately one third of patients with sporadic LAM and 80–90% of patients with TSC-LAM.34

Lymphangioleiomyomatosis. Computed tomography of the chest showing multiple, well-defined, round, thin-walled cysts scattered throughout both lungs.
Summary of Clinical, Radiographic, and Pathologic Features of Common DCLDs
Pathology
Histologic diagnosis of LAM relies on the presence of 2 key features: cystic change and LAM cells (Fig. 2). In early disease stages, the cysts and LAM cells may be inconspicuous, highlighting the need to correlate the pathology with clinical and radiologic findings. Small nests of LAM cells are typically located at the periphery of cysts. In more advanced disease, LAM cells form small nodules and infiltrate small airways and vessel walls, leading to vascular destruction and accumulation of hemosiderin-laden macrophages. LAM cell clusters, composed of a LAM cell core surrounded by an outer lining of lymphatic endothelial cells, can be found within lymphatic channels in the lung as well as in chylous effusions, thus representing a mechanism for LAM cell dissemination.36 LAM cells are morphologically heterogeneous, consisting of haphazardly arranged bundles of spindle cells and cuboidal to oval epithelioid cells. LAM cells have a characteristic immunophenotype that is diffusely positive for smooth muscle actin and vimentin, with a subpopulation of LAM cells staining positively with the human melanoma black-45 (HMB-45) antibody developed against glycoprotein-10, a premelanosomal protein.37 LAM cells also stain positive for estrogen and progesterone receptors.38

Histopathology of lymphangioleiomyomatosis. (A) Multiple cystic spaces are seen at low magnification (*) (40×). (B) Higher magnification of cyst (*) wall showing a lymphangioleiomyomatosis (LAM) cell bundle composed of aggregates of bland, elongated, spindled cells (arrow) and round to oval epithelioid cells (arrowheads), some with clear cytoplasm (open arrowhead) (600×). (C) Hemosiderin is often present within alveolar macrophages in the air spaces (arrow) as well as in the interstitium (arrowhead) (600×). (D) The LAM cells (arrows) within the cyst (*) walls are positive for smooth muscle actin (SMA) (400×). (E) SMA (brown stain) with a subpopulation of cells staining positively for the melanocytic marker, HMB-45 (400×). (F) Brown stain and arrow indicates cells staining positively for HMB-45 (400×).
Diagnosis
The diagnosis of LAM should be considered in any woman who presents with unexplained progressive dyspnea or spontaneous pneumothorax.39–41 In women with a known diagnosis of TSC, it is recommended that screening HRCT be performed starting at the age of 18 y to look for the presence of LAM.42 The presence of characteristic cystic change on HRCT in conjunction with TSC is considered sufficient to establish a clinical diagnosis of LAM.43,44 In patients with suspected sporadic LAM, it is essential to perform a detailed physical examination to evaluate for the presence of underlying TSC because adult-onset diagnosis of TSC is not uncommon.6 Serum VEGF-D levels are elevated in ∼ 70% of patients with sporadic LAM and are nearly 100% specific for the diagnosis of LAM in patients with cystic lung disease on HRCT. In the United States, serum VEGF-D levels can be obtained in a College of American Pathologists/Clinical Laboratory Improvement Amendments approved manner at the Translational Trial Development and Support Laboratory (TTDSL; available at: https://www.cincinnatichildrens.org/research/cores/translational-core-laboratory/translational-trial-development-support-laboratory. Accessed September 2, 2019) and should be obtained prior to performing invasive diagnostic procedures.44–46 Renal AMLs can be seen in about one third of patients with sporadic LAM and can help establish diagnosis of LAM without needing a lung biopsy. Similarly, other lymphatic manifestations such as chylous fluid collections and lymphangioleiomyomas can be seen in ∼ 20% of sporadic LAM patients and obviate the need for lung biopsy. In total, the diagnosis of LAM can be established with the above-mentioned noninvasive measures in 70–80% of patients. In the absence of one of the above-mentioned criteria, lung biopsy may be required to confirm the diagnosis of LAM. It is worth highlighting that, in certain situations, especially patients with mild disease who would not warrant initiation of pharmacotherapy, a probable diagnosis of LAM with serial monitoring may be a reasonable strategy. In cases where a definite diagnosis is needed, transbronchial lung biopsy has a yield of > 60%, appears to be safe based on small series,47–49 and should be performed prior to pursuing surgical lung biopsy.50 Critical review of the pathology by an expert pathologist is essential to avoid false negatives on biopsy. An algorithmic approach to establishing the diagnosis of LAM has recently been published.50
Management
Improved understanding of the pathobiology of LAM has led to rapid advancements in disease management. In May 2015, the U.S. Food and Drug Administration (FDA) approved sirolimus, an mTOR inhibitor for the treatment of LAM. The approval was based largely on the findings of the Multi-center International LAM Efficacy of Sirolimus (MILES) trial, a double-blind, randomized, parallel-group trial of 1 y of treatment with sirolimus versus placebo, followed by 1 y of observation. Subjects who were treated with placebo lost ∼ 10% of their lung function over the course of the treatment year, whereas subjects who received sirolimus had stable lung function and improved functional performance.51 The recently published American Thoracic Society/Japanese Respiratory Society LAM Clinical Practice Guidelines recommend that sirolimus be initiated for patients with LAM who have FEV1 ≤ 70% of predicted, problematic chylous effusions, or rapidly progressive disease or substantial disease burden.44 Because the effect of the drug is suppressive rather than curative, most patients are maintained on treatment indefinitely. In the clinical experience that has accumulated since the publication of MILES results, sirolimus appears to have durable safety and efficacy, and a majority of the LAM patients tend to exhibit a beneficial response to treatment with sirolimus.
Further trials are underway to assess the risks and benefits of treating patients in early disease stages (MILED, NCT03150914) and to elucidate the ideal dose and duration of therapy. Several early-stage clinical trials involving novel agents, either alone or in combination with sirolimus, are either underway or have recently been completed: aromatase inhibitors (NCT01353209),52 combined hydroxychloroquine and sirolimus (NCT01687179),53 combination of sirolimus and simvastatin (NCT02061397), combined resveratrol and sirolimus (NCT03253913), celecoxib (NCT02484664), imatinib (NCT03131999), and nintedanib (NCT03062943). We are optimistic that results from these studies will help further refine management of LAM in the near future.
Given the high risk of pneumothorax recurrence in LAM patients (ipsilateral recurrence > 70%), pleurodesis should be performed after the first pneumothorax episode.54 Ipsilateral recurrence rate after pleurodesis is reduced by ∼ 50% and ranges between 27% and 32% (Table 4).54 It is important to note that prior pleurodesis is not a contraindication for future lung transplantation.50,55 Patients with LAM should be counseled to avoid smoking (Table 5). Exogenous estrogen can accelerate disease progression in LAM, and patients should be counseled to avoid hormonal therapies containing estrogen. LAM patients should stay up to date with their vaccinations, including influenza and pneumococcal vaccines. Finally, lung transplantation remains a viable option for patients with progressive end-stage LAM that does not respond to other therapies.56,57 Recommendations for the optimal diagnosis and management of LAM have been published43,44,50 and are a valuable resource for clinicians to guide effective decision making when evaluating patients with suspected or confirmed LAM.
Pneumothorax in DCLDs
General Recommendations Applicable to All Patients With DCLDs
Prognosis
Several prognostic biomarkers in the progression of LAM have been implicated in previous studies, including baseline PFT values,24 menopausal status,24 symptomatology at presentation,58 CT findings,59 association with TSC,59 response to bronchodilators,60 and VEGF-D levels.61 A recently published longitudinal analysis of 217 LAM patients enrolled in the National Heart, Lung and Blood Institute LAM Registry has shown 5-, 10-, 15-, and 20-y transplant-free survival rates of 94%, 85%, 75%, and 64%, respectively, with a median survival > 20 y. Baseline PFTs and menopausal status were associated with future risk of progression to death or transplantation.27 Similar results with regard to the impact of menopausal status on the rate of disease progression were also noted in a recent post hoc analysis of the MILES cohort.62
Pulmonary Langerhans Cell Histiocytosis
Pulmonary Langerhans cell histiocytosis (PLCH) is a rare DCLD that is thought to be caused by tobacco smoking because it almost exclusively affects current or previous smokers.63 In some cases, PLCH might present as a multisystem disease involving other organs, most commonly bones and pituitary.
Pathophysiology
PLCH was previously thought to occur as a result of polyclonal proliferation of Langerhans cells induced by cigarette smoke; differentiated subpopulation of macrophages that regulates mucosal airway immunity.64,65 However, pathogenic activating mutations in the mitogen-activating protein (MAP) kinase pathway, especially BRAF V600E, have recently been described in > 50% of the lesional dendritic cells in subjects with PLCH.66 This discovery has transformed our understanding of the pathogenesis of PLCH, and it is now considered to be an inflammatory myeloid neoplasm, where cigarette smoke induces the abnormal proliferation and migration of MAP kinase–mutated dendritic cells to the lungs, leading to the development of PLCH. Secondary activation of the immune system and subsequent destruction of the bronchiolar walls by matrix-degrading enzymes also plays a key role in the development of the hallmark peribronchiolar nodules, cavities, and eventual cystic destruction.5
Clinical Presentation
Patients usually present with cough, dyspnea, pleuritic chest pain, and occasionally hemoptysis or spontaneous pneumothorax. Many patients with PLCH remain asymptomatic, and PLCH findings are noted incidentally on chest imaging.67 In early stages of the disease, PFTs may be normal or exhibit mild restriction; in later stages, obstructive defects are common, along with air trapping and reduction in DLCO.67 In cases of multisystemic Langerhans cell histiocytosis, patients might present with skin rash, lytic bone lesions, or diabetes insipidus.
Radiology
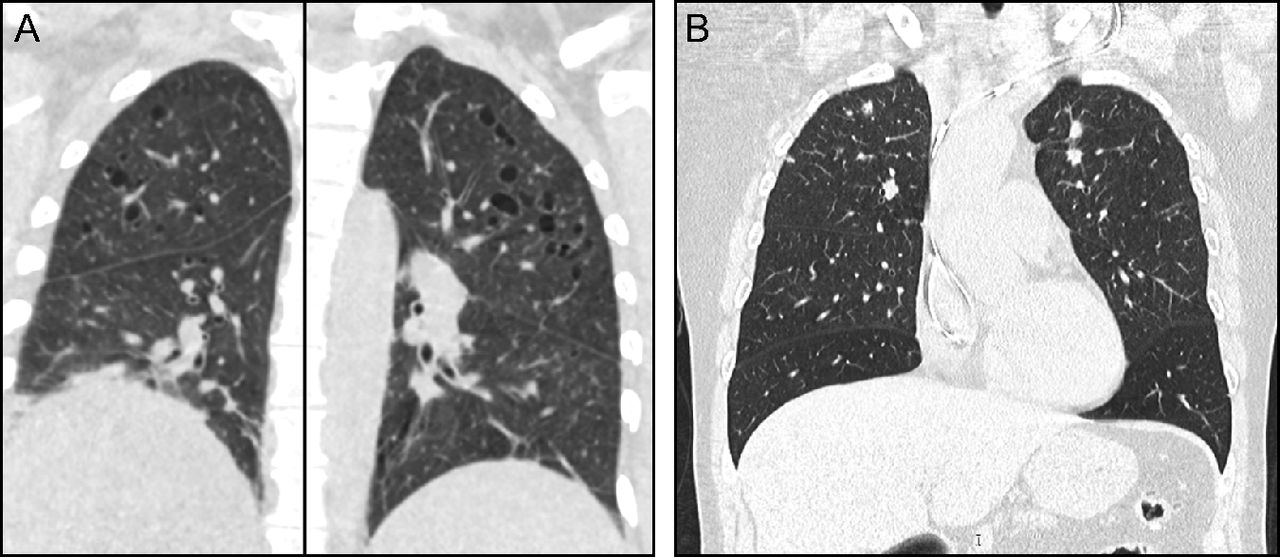
Chest CT findings typically include a combination of nodules and cysts. PLCH nodules are usually 1–10 mm in size and are typically centered around the bronchioles. Cysts in PLCH usually appear in later stages of the disease, can vary in wall thickness from typical thin-walled cysts to thick-walled cavities, and tend to spare the bases of the lungs (Fig. 3, Table 3). Other radiologic findings emanating from concomitant cigarette smoke-induced lung damage such as emphysema, ground-glass opacities suggestive of respiratory bronchiolitis/Desquamative interstitial pneumonia, or reticular opacities are occasionally seen in patients with PLCH.68 PLCH lesions, especially nodules, can be fluorodeoxyglucose (FDG)-avid, making it difficult to differentiate between PLCH and other malignant conditions on the basis of FDG-positron emission tomography.69
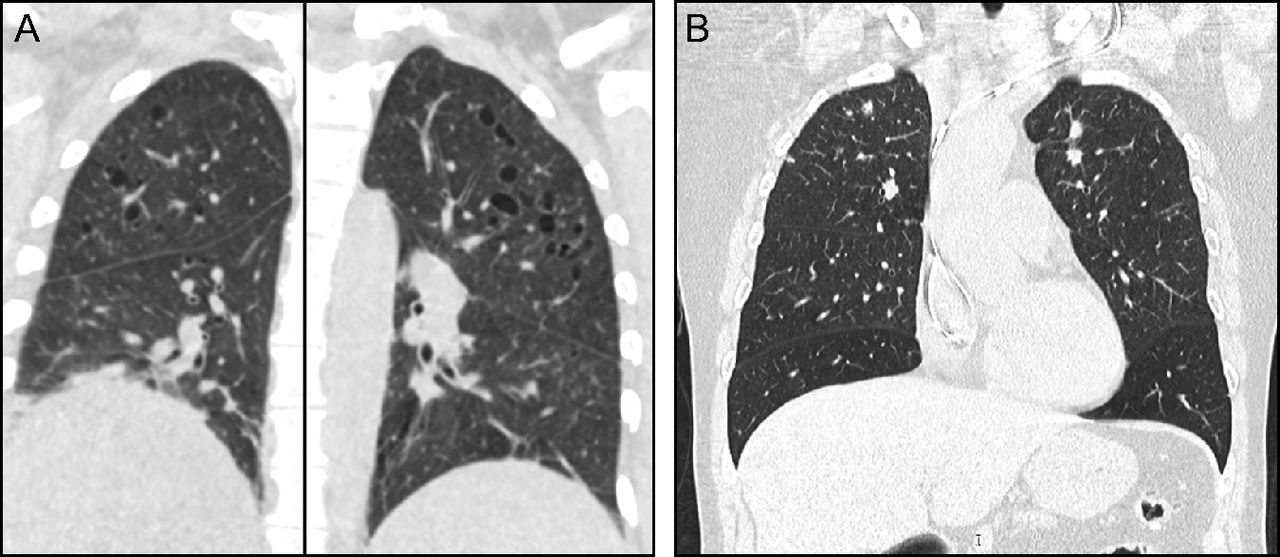
Pulmonary Langerhans cell histiocytosis (PLCH). Computed tomography of the chest typically shows a combination of nodules and cysts predominantly seen in upper and middle lung zones with characteristic costophrenic angle sparing. (A) Cystic predominant PLCH. (B) Nodular predominant PLCH.
Pathology
The classic histopathology for PLCH is a patchy, peribronchiolar nodular infiltrate composed of clusters of dendritic cells admixed with variable numbers of other cell types including lymphocytes, macrophages, and eosinophils (Fig. 4). Central cavities can form within the nodules, which may represent dilation of the former bronchiolar lumen. The inflammatory infiltrate extends into the surrounding alveolar septa, resulting in the characteristic stellate configuration. Fibrosis with collagen deposition can also be present within the nodules, leading to traction and enlargement of the surrounding air spaces. In advanced PLCH, fibrosis may be the predominant component with only few dendritic cell aggregates at the periphery or only a stellate scar suggesting the diagnosis of PLCH. The key diagnostic feature of PLCH is the presence of Langerhans-like cell clusters. Scattered interstitial Langerhans-like cells that are common in normal lungs and can be increased in association with smoking are not diagnostic of PLCH. Pathogenic Langerhans-like cells in PLCH are characterized as bland appearing cells with a moderate amount of eosinophilic cytoplasm and distinctive folded or kidney-shaped nuclei. The cells stain positively for S100 protein, Langerin, and CD1a.70 Histopathologic features of other smoking-related lung diseases are commonly seen in PLCH, including respiratory bronchiolitis, Desquamative interstitial pneumonia and smoking-related interstitial fibrosis.

Nodular lesion in patient with Langerhans cell histiocytosis. (A) Well circumscribed nodular infiltrate (arrow) with infiltrate extending into the adjacent alveolar septa (arrowhead) producing the characteristic stellate configuration. Some of the surrounding air spaces are enlarged (*) due to traction caused by fibrosis within the lesion (20×). (B) High magnification of diagnostic aggregates of Langerhans cells with eosinophilic cytoplasm and characteristic folded and kidney bean shaped nuclei (arrows). Pigmented macrophages associated with cigarette smoking (arrowheads) are frequently found within the inflammatory infiltrate and in surrounding alveolar spaces (1,000×). (C) The Langerhans cells stain positively for S-100, Langerin, and CD1a (arrow). The Langerhans cells in lesions with a mixed inflammatory infiltrate and background fibrosis with collagen deposition as in this case are often most prominent in the interstitium at the periphery of the nodule (40×).
Diagnosis
In some cases, the diagnosis of PLCH might be sufficiently made on the basis of a CT scan if all of the classic pulmonary findings are present along with history of smoking. In other cases where the diagnosis is questionable, further tests might be required, including bronchoscopy with bronchoalveolar lavage and biopsy. Although it has a high false negative rate, bronchoalveolar lavage showing > 5% CD1A+ cells is strongly suggestive of PLCH. A more definitive diagnosis requires tissue confirmation either via transbronchial or surgical lung biopsy.47,71
Management
The mainstay of PLCH management is smoking cessation, with reports of disease stabilization and even regression reported after smoking cessation.72–74 However, a subset of patients with PLCH (∼ 30%) continue to decline even after successful smoking cessation. The inability to prospectively identify this subset highlights the importance of close, longitudinal PFT monitoring for patients with PLCH.75 Multiple chemotherapeutic regimens have been tried in patients with PLCH with limited success. Currently, the most promising treatment approaches include cladribine76,77 and targeted treatment aimed at the MAP kinase pathway.78 About 20% of patients with PLCH experience a spontaneous pneumothorax (Table 4). The risk of recurrence is high (∼ 60%) if managed conservatively,79 thus patients with PLCH should undergo pleurodesis following the first episode of spontaneous pneumothorax rather than wait for a recurrent event and undergo periodic evaluation for the development of pulmonary hypertension. Management of PLCH-associated pulmonary hypertension can be difficult and best performed at expert referral centers. Lung transplantation is a viable option in advanced disease.80
Prognosis
The long-term prognosis and overall survival of patients with PLCH is not well established; however, retrospective case series have reported median survival times of 12.5–13 y from the time of diagnosis.67,81 The development of pulmonary hypertension in patients with PLCH has been associated with worse outcomes.82,83 Other reported factors associated with poor outcomes include older age, low FEV1, reduced FEV1/FVC, high residual volume, and reduced DLCO.67,81 Continued smoking has a deleterious impact on the rate of disease progression, and successful smoking cessation has been shown to reduce the rate of disease progression.84 Further clarity regarding the natural history of disease progression in PLCH and identification of biomarkers that can help predict the subset of patients at increased risk of disease progression remains a future research priority for PLCH.
Birt-Hogg-Dubé Syndrome
Birt-Hogg-Dubé Syndrome (BHD) is a rare autosomal-dominant disease that is characterized by lung cysts, hair follicle tumors, and renal tumors. Lung cysts from BHD are usually seen in the fourth decade of life, with 80% penetrance by the age of 50 y.85,86
Pathophysiology
BHD is caused by mutations in the Folliculin gene (FLCN), which encodes folliculin, a tumor-suppressor protein. The exact pathophysiology of pulmonary cyst formation in BHD is unclear; some of the candidate pathways that have been proposed in the pathogenesis of pulmonary cystogenesis include the mTOR signaling pathway, E-cadherin-LKBP1-AMPK signaling, increased cell-cell adhesion, and increased mechanical stress on the alveolar-septal regions in the anatomically weak lung areas.87–90
Clinical Presentation
Pulmonary involvement from BHD can be detected either incidentally or following a spontaneous pneumothorax. The prevalence of spontaneous pneumothorax in patients with BHD has been variably reported to range between 25% and 75%.86,91,92 Spontaneous pneumothorax in patients with BHD has been reported in the absence of radiologically visible cysts on chest CT.93 PFTs generally show preserved spirometric parameters including FEV1, FVC, and FEV1/FVC, and a mild reduction in DLCO is seen in some advanced cases.94 Skin lesions including fibrofolliculomas and trichodiscomas are the most common manifestation of BHD, found in ∼ 90% of the patients.91,95 Typical BHD skin lesions are whitish, dome-shaped papules that measure 1–5 mm in size and are usually seen on the facies and upper torso. Renal tumors are seen in up to 27% of patients with BHD (oncocytomas and chromophobe renal carcinoma are the most common), who have an average age of 50 y at diagnosis.96
Radiology and Pathology
Pulmonary cysts are present in ∼ 80% of the adult patients with BHD. BHD lung cysts classically present as multiple, thin-walled cysts that range widely in size, are variable in shape (round to lentiform), and are usually found at the bases, often abutting the pleural surface and pulmonary vasculature (Fig. 5, Table 3).97 BHD lung cysts are predominantly subpleural or paraseptal in location by histologic evaluation and are surrounded by normal thin alveolar walls without associated cellular proliferations, significant inflammation, or fibrosis (Fig. 6). The cysts can be difficult to distinguish from emphysema based on histopathology; however, in contrast to smoking-related emphysema, BHD cysts predominantly occur in the lower rather than upper lobes, and the surrounding parenchyma can be compressed but is characteristically normal. Additionally, in a study of 229 pulmonary cysts in 50 subjects with BHD, 88.2% of BHD lung cysts abutted interlobular septa and 13.6% were found to have intracystic septa with occasional venules protruding into the cysts.87 These features along with the location of the cysts in the lower lobes, the lack of significant inflammation and fibrosis, and the absence of cellular proliferations are key characteristics that distinguish BHD from other DCLDs. The diagnosis of BHD should thus be considered in any case with multiple, lower lobe-predominant, nondescript cysts, especially in nonsmokers.
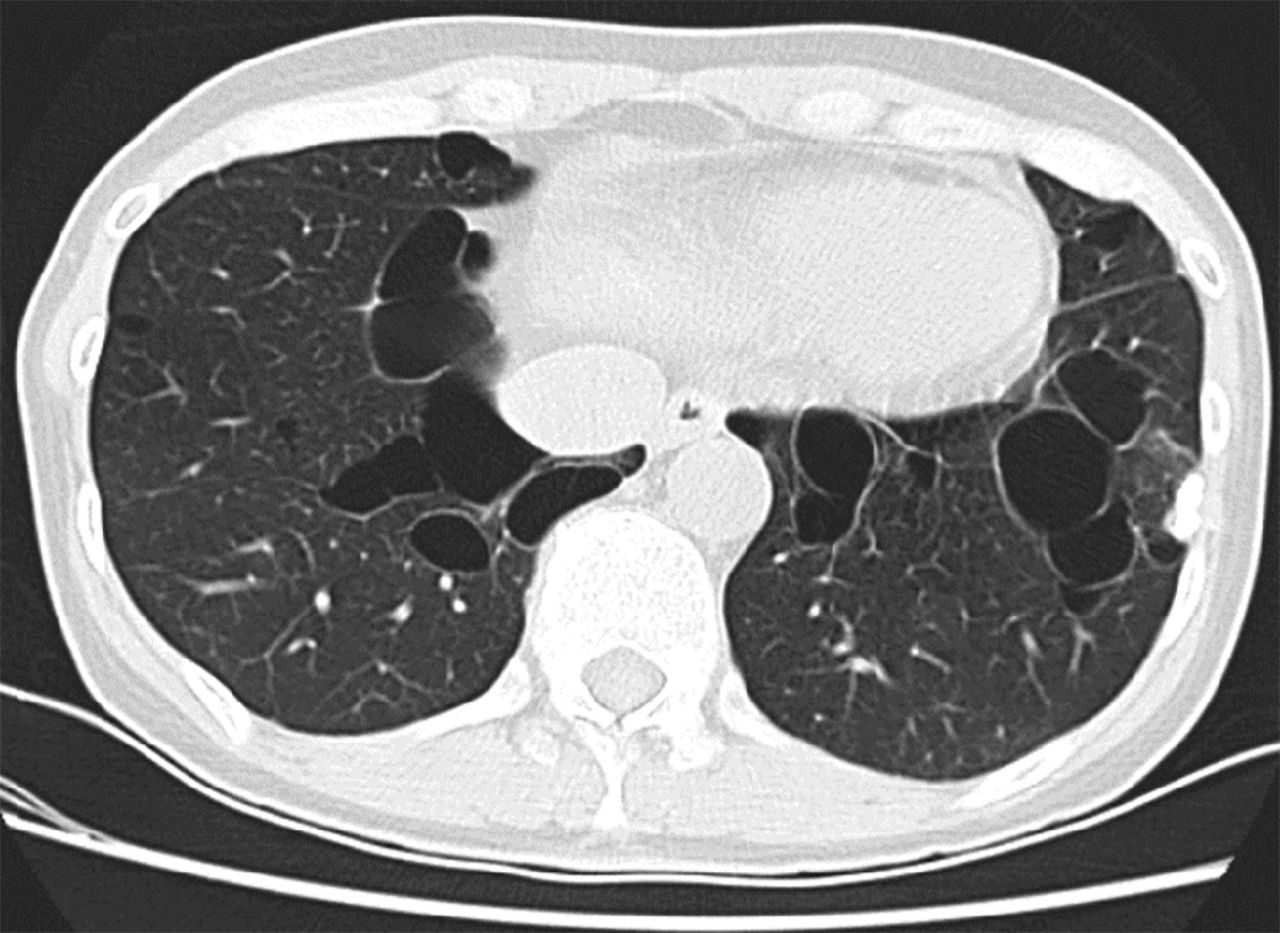
In Birt-Hogg-Dubé syndrome (BHD), lung cysts can range widely in size, often have an oval or lentiform shape, and are usually found at the lung bases, often abutting the pleura or pulmonary vasculature.
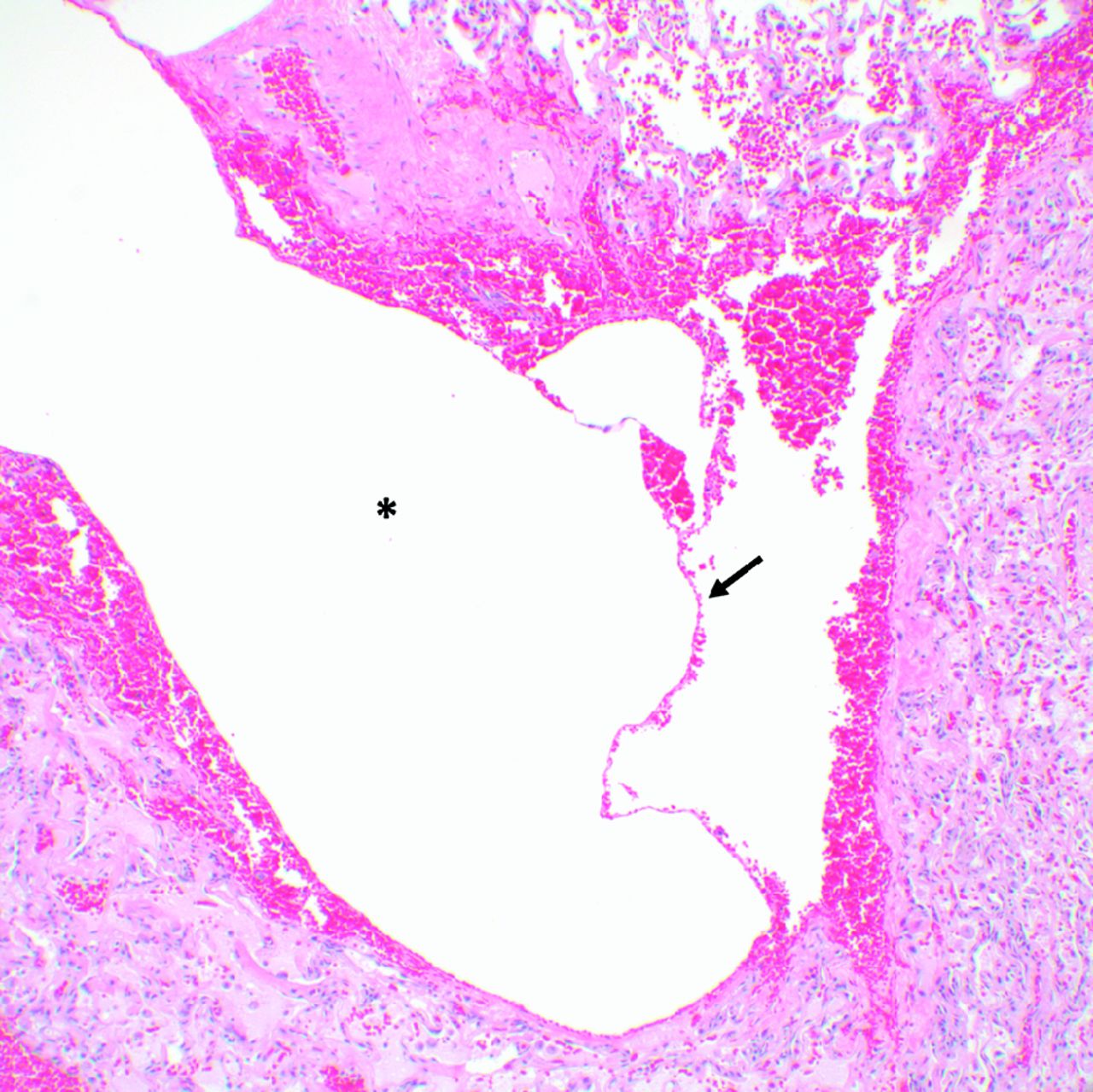
Lung cyst from a patient with Birt-Hogg-Dubé syndrome. A paraseptal cyst (*) surrounded by compressed lung parenchyma without significant inflammation, fibrosis, or cellular proliferations. A thin intracystic septum (arrow) traverses the cyst (100×).
Diagnosis
Young patients who present with spontaneous pneumothorax along with a personal or family history of skin lesions, pneumothorax, or renal cancer should be evaluated for BHD. Careful skin examination should be performed for all patients with suspected BHD, with a low threshold for dermatological evaluation and skin biopsy of suspicious lesions, especially the lesions in characteristic places such as the face and upper torso. Detection of fibrofolliculomas on skin biopsy in conjunction with typical chest CT findings can be sufficient to make a clinical diagnosis of BHD. It is important to note, however, that there is wide phenotypic variability in the clinical manifestations of BHD, and the absence of skin or kidney lesions does not preclude the possibility of underlying BHD. Detection of pathogenic FLCN mutations from the peripheral blood is required to establish a confirmed genetic diagnosis of BHD. Given the high penetrance of FLCN gene mutations, screening asymptomatic family members is recommended. Diagnostic criteria for BHD have previously been published by us and by others.98,99
Management and Prognosis
Patients should be reassured that cysts from BHD typically occupy a small percentage of the lung parenchyma, do not cause physiological impairment, and tend to stay stable over time. Management of pulmonary involvement from BHD is typically centered on the increased risk of spontaneous pneumothoraces. Although the exact prevalence of spontaneous pneumothorax in BHD has been variably reported to range between 25% and 75%,86,100 there is an extremely high risk of recurrence (∼ 75%) if managed conservatively (Table 4).86,100 Thus, patients with BHD presenting with a spontaneous pneumothorax should undergo pleurodesis following the first episode of pneumothorax rather than waiting for a recurrent episode. Patients with BHD should be screened regularly for renal tumors, starting at age 21 y. Magnetic resonance imaging is the best modality for screening due to the risk of cumulative radiation exposure from CT scans and the lack of sensitivity for detecting small lesions with ultrasound.101 In patients with no detectable lesion on the screening scan, further imaging can be performed every three years. More frequent imaging may be needed in patients with kidney lesion(s), with the frequency dictated by the size, morphology, and imaging characteristics of the underlying lesion(s).
Cystic Lung Diseases Associated With Lymphoproliferative Disorders
Lymphoid Interstitial Pneumonia/Follicular Bronchiolitis
Lymphoid interstitial pneumonia (LIP) is a form of interstitial lung disease that results from lymphocytic infiltration of the lung parenchyma.102 Follicular bronchiolitis (FB) is characterized by hyperplasia of lymphoid follicles along the walls of bronchioles and blood vessels consistent with a lymphatic distribution.103 The majority of LIP and FB cases are associated with rheumatologic disorders (especially Sjögren syndrome, systemic lupus erythematosus, and rheumatoid arthritis), immunodeficiencies (including common variable immunodeficiency), and viral infections (like Epstein-Barr virus and human immunodeficiency virus).104
Pathophysiology and Clinical Presentation.
LIP and FB constitute a pathophysiologic spectrum of lymphocytic infiltration from hyperplasia of bronchus-associated lymphoid tissue. The association of LIP and FB with rheumatologic disorders and immunodeficiencies suggests that bronchus-associated lymphoid tissue hyperplasia might represent cellular response to a variety of intrinsic and extrinsic stimuli.103 Patients with LIP and FB usually present with nonspecific symptoms including cough, dyspnea, fatigue, fever, and weight loss. PFTs typically show a restrictive pattern with reduced DLCO.104 Because most cases of LIP and FB are associated with underlying connective tissue diseases, these patients may also have other symptoms including rash, sicca symptoms, Raynaud's, and other findings of individual connective tissue diseases.
Radiology and Pathology.
Chest CT might show findings of ground glass opacities and centrilobular and subpleural nodules in addition to lung cysts. Cysts in LIP usually have a diffuse random distribution and often contain internal structures and eccentric vessels that can help distinguish LIP from other forms of DCLDs (Fig. 7).105,106 Tissue examination in LIP shows diffuse infiltration of alveolar septa by a mixture of small lymphocytes, plasma cells, and macrophages, typically with little or no fibrosis. Lymphoid follicles with germinal centers are often present. The lymphocytes are predominantly T cells with scattered B cells except within the germinal centers, where B cells are prominent.102 Alveolar spaces may be distorted and contain proteinaceous material along with scattered lymphocytes and macrophages.107 Occasionally loose granulomas are present, but well-formed granulomas are not seen.107 In FB, numerous lymphoid follicles with reactive germinal centers are seen in a peribronchial and peribronchiolar distribution103 (Fig. 8). Bronchiolar lumens are often narrowed by the inflammatory nodules. In some cases, the cellular infiltrate extends beyond the peribronchiolar interstitium, involving the alveolar septa focally rather than the diffuse involvement diagnostic of LIP, which supports the concept that FB and LIP represent entities along a shared disease spectrum.102,103 Careful histopathologic evaluation is important to rule out other lymphoid disorders including lymphoma. Flow cytometry, immunohistochemical staining, and analysis for clonality are helpful for this purpose102,107

Lymphoid interstitial pneumonia (LIP) in a patient with Sjögren syndrome. Cysts in LIP usually have diffuse random distribution, round or oval shape, and are variable in size, and they are frequently are associated with eccentric vessels and intracystic septations or structures.
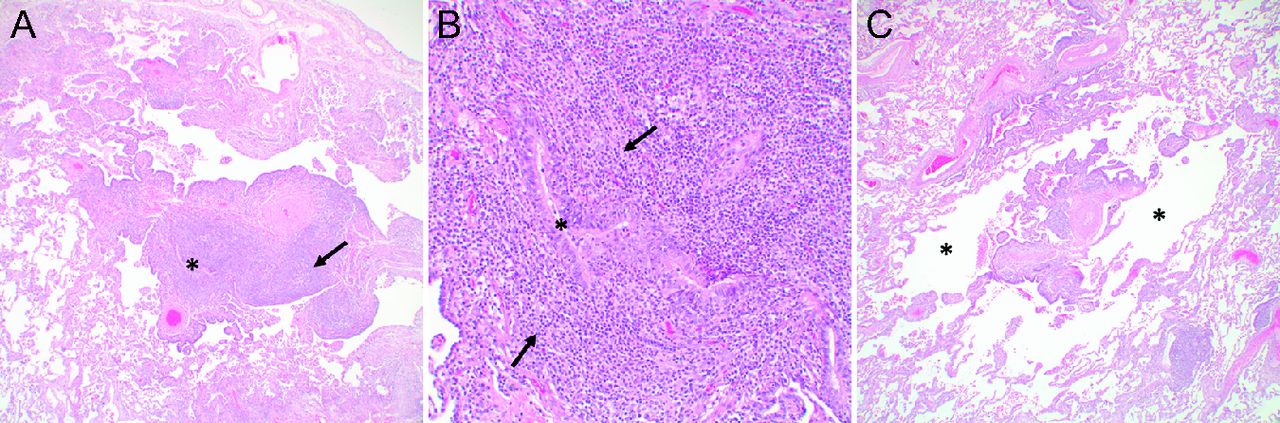
Follicular bronchiolitis and cystic spaces in a patient with Sjögren syndrome. (A) Dense lymphoid infiltrate with germinal centers (arrow) surrounding a bronchiole (*) (40×). (B) Higher magnification of the bronchiole in (A) showing narrowing of the bronchiolar lumen by the prominent chronic inflammatory infiltrate (arrows) (200×). (C) Markedly inflamed bronchiole with cystically dilated air spaces (*) surrounding the bronchiole (40×).
Diagnosis.
The diagnosis of LIP and FB can be established on clinical grounds in patients with characteristic cystic changes on HRCT and the presence of underlying autoimmune disease such as Sjögren syndrome.106 In some cases, however, cystic changes from LIP/FB can be the presenting manifestation of underlying Sjögren syndrome, and the serologies may be falsely negative. In unclear cases, surgical lung biopsy may be needed to establish a histopathological diagnosis.104
Management.
LIP is usually treated with steroids and cytotoxic agents. The ground glass attenuation and nodules associated with LIP respond well to immune suppression, but the effect of these treatments on cystic changes is unclear.108 LIP secondary to human immunodeficiency virus infection may improve with initiation of highly active antiretroviral therapy.109 There is an increased risk of development of lymphoma, and repeat chest imaging should be conducted periodically (eg, every 3–5 y) in these patients; providers should maintain a low threshold for biopsy in case of new or enlarging nodules or consolidative opacities.
Amyloidosis
Amyloidosis refers to a group of disorders characterized by abnormal deposition of amyloid fibrils. This could present as a systemic disorder or as a localized disorder. Localized pulmonary amyloidosis usually presents with pulmonary nodules, but in rare cases it may present with diffuse cystic changes.110 Lung amyloidosis could be seen with Sjögren syndrome, which might be associated with mucosa-associated lymphoid tissue lymphoma in some cases.111 Diagnosis of pulmonary amyloidosis requires histopathological examination by lung biopsy. Amyloid material is typified as globular irregular deposits of amorphous eosinophilic glassy material on routine histologic stain. A collection of macrophages known as foreign-body giant cell response to the amyloid is commonly present along with occasional calcification or ossification. Chronic inflammation may be present with clusters of plasma cells with monoclonal staining for kappa or lambda immunoglobulin light chains. Congo red staining with apple green birefringence under polarized light, immunohistochemical staining for amyloid proteins, or electron microscopy findings of thin amyloid fibrils can aid in the diagnosis. Liquid chromatography and tandem mass spectrophotometry can be used to subtype the amyloid.
Light-Chain Deposition Disease (LCDD)
Light-chain deposition disease (LCDD) is a rare disease that is characterized by deposition of a non-fibrillary amorphous material in alveolar walls and small airways. Contrary to amyloidosis, this acellular material does not have β-pleated sheet configuration and therefore does not bind Congo red stain.112 In rare instances, LCDD can have isolated pulmonary involvement; however, LCDD is usually associated with multiple myeloma and other lymphoproliferative disorders, and renal involvement resulting in proteinuria is common among patients.113 LCDD lung cysts vary in size and shape and could resemble LAM or PLCH on chest radiography.114 LCDD is usually a progressive disorder that results in respiratory failure. Treatment involves treating the underlying lymphoproliferative disorder (if present), with lung transplantation being an option for advanced cases.114–116
Summary
DCLD on chest imaging carries a broad differential diagnosis and can occur as a result of multiple pathophysiologically distinct disease processes. The presence of cysts in the pulmonary parenchyma creates unique physiological and clinical implications for respiratory providers, including respiratory therapists (Table 6). Establishing the correct diagnosis is crucial because DCLDs vary widely in clinical course, prognosis and treatment. Chest HRCT remains the most important noninvasive tool for evaluation of DCLDs. Careful and systematic evaluation of the cyst characteristics on HRCT, integrated with clinical, laboratory, and histopathology (if available) features can help narrow the field and guide the clinician toward the right path in the diagnosis and management of these patients.
Respiratory Care Implications for DCLD Patients
Footnotes
- Correspondence: Nishant Gupta MD, 231 Albert Sabin Way, MSB Room 6053, ML 0564, Cincinnati, OH 45267. E-mail: guptans{at}ucmail.uc.edu.
- Copyright © 2020 by Daedalus Enterprises
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.
- 30.↵
- 31.↵
- 32.↵
- 33.
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.
- 46.↵
- 47.↵
- 48.
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.
- 89.
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 113.↵
- 114.↵
- 115.
- 116.↵
- 117.
- 118.
- 119.
- 120.
- 121.
- 122.
- 123.
- 124.
- 125.
- 126.
- 127.
- 128.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}