

Abstract
Contemplating the future should be grounded in history. The rise of post-polio ICUs was inextricably related to mechanical ventilation. Critically ill patients who developed acute respiratory failure often had “congestive atelectasis” (ie, a term used to describe ARDS prior to 1967). Initial mechanical ventilation strategies for treating this condition and others inadvertently led to ventilator-induced lung injury. Both injurious ventilation and later use of overly cautious weaning practices resulted from both limited technology and understanding of ARDS and other aspects of critical illness. The resulting misperceptions, misconceptions, and missed opportunities took decades to rectify and in some instances still persist. This suggests a reluctance to acknowledge that all therapeutic strategies reflect the historical period in which they were developed and the corresponding limited understanding of ARDS pathophysiology at that time. We are at the threshold of a revolutionary moment in critical care. The confluence of enormous clinical data production, massive computing power, advances in understanding the biomolecular and genetic aspects of critical illness, and the emergence of neural networks will have enormous impact on how critical care is practiced in the decades to come. Therefore, it is imperative we understand the long-crooked path needed to reach the era of protective ventilation in order to avoid similar mistakes moving forward. The emerging era is as difficult to fathom as our current practices and technologies were to those practicing 60 years ago. This review explores the history of mechanical ventilation in treating ARDS, describes current protective ventilation strategies, and speculates how ARDS management might look 20 years from now.
- ARDS
- ventilator-induced lung injury
- lung-protective ventilation
- right heart–protective ventilation
- diaphragmatic protective ventilation
- recruitment maneuvers
Introduction
“You can’t really know where you are going until you know where you have been.”
Maya Angelou1
“The past is never dead. It's not even past. … Haunted by wrong turns and roads not taken, we pursue images perceived as new but whose providence dates to the dim dramas … which are themselves but ripples of consequence echoing down the generations.”
William Faulkner2
I am deeply honored to deliver the first scientific memorial lecture for my colleague and friend Bob Kacmarek and to speak on a subject I know Bob would have relished delivering himself. When I began thinking about this topic, what came to mind was something the poet Maya Angelou said. I believe speculating about the future without discussing what has transpired over the past 60 years would be of marginal value. And toward completion of this project I was reminded of a passage by novelist William Faulkner. Although written in a different context nonetheless it captures something of the crooked path taken to reach the current era of protective ventilation.
In no small measure, respiratory care arose from the rapid expansion of ICUs in the early to mid-1960s, largely driven by the need to safely deliver mechanical ventilation. Bob and I entered respiratory care during what could be called the “prehistoric period” of ICU mechanical ventilation (1965–1975). There was no history because it was all new and happening while we were doing it. History helps us reflect upon why specific approaches came about and subsequently were abandoned, retained, and sometimes rediscovered. Reckoning with our past allows us to appreciate all-too-human tendencies to misperceive, misconceive, and miss opportunities that have occurred along the way.
I believe we stand at the threshold of a truly revolutionary era in our understanding and management of ARDS. One that will challenge our profession to adapt moving deeper into the 21st century. Therefore, it’s imperative we recognize the tenacious hold outdated approaches and beliefs have had and (in some cases) continue to have. Thus preparing us for what lies ahead. I will also review the current state and scope of protective ventilation practices in ARDS before attempting to answer the question “whither goest thou?”
Origins of Ventilator-Induced Lung Injury
Absorption Atelectasis, Large Tidal Volumes, and Acute Respiratory Failure
In the post-polio period of 1958–1961, small ICUs with 5–6 bed capacities emerged in major university-associated hospitals (eg, Oxford, Johns Hopkins, Harvard, and the universities of Toronto, Pittsburgh, and Southern California).3,4 By the mid-1960s, ICUs had expanded widely in Western countries. During this period, the indications for mechanical ventilation also expanded to treat acute respiratory failure across a wide array of medical, surgical, and trauma cases. Beginning in 1963, these early experiences began to appear in the medical literature highlighting the problems encountered in managing acute respiratory failure.5-7 These patients differed from those with polio who were characterized by respiratory muscle paralysis, normal lung mechanics and gas exchange, low minute ventilation (V̇E) demand, and in whom triggering and asynchrony were essentially nonfactors.
The customary practice of supine positioning with mechanical ventilation using atmospheric expiratory pressure and a physiologic tidal volume (VT) based upon the Radford nomogram (developed for use during general anesthesia and in managing patients with polio)8 resulted in progressive atelectasis and hypoxemia from intrapulmonary shunting. The predominant clinical model for treating hypoxemia was that based upon the reversal of postoperative absorption atelectasis.7 This involved sustained (“sigh”) inflations of 30–40 cm H2O for 15 s that was adopted for ICU practice.
For practical reasons, the sustained inflation approach was modified by simply using large VT ventilation to treat atelectasis, hypoxemia, and also to meet elevated V̇E demands.7 The later justification was that the Radford nomogram grossly underestimated V̇E requirements as physiologic dead-space and CO2 production are markedly elevated in acute respiratory failure (as predicted by the nomogram’s authors).5,8 And despite the use of predicted normal VT, patients often complained of dyspnea or appeared in distress that was quickly relieved once VT was increased.3 In addition, using positive pressure ventilators designed to treat patients with polio was inadequate as they could only achieve peak airway pressures of 15–20 cm H2O.7
By the mid-1960s, a mean VT of ∼11–13 mL/kg with ambient expiratory pressure was the standard of care,5,6 as had been recommended.7 And in patients presenting with (or subsequently developing) severe acute respiratory failure, the only perceived options were to increase VT and FIO2 concentration to toxic levels (ie, ≥ 0.70).9 Moreover, in subjects who now would be classified as primary ARDS, increasing VT to ≥ 15 mL/kg increased alveolar ventilation but was ineffective in reversing hypoxemia.6
Thus, the absorption atelectasis paradigm supporting high VT ventilation for refractory hypoxemia was not conceptually appropriate. And during this period, it was also discovered that the Venturi air-entrainment mechanisms used to control FIO2 at 0.40 were deeply flawed. When peak inspiratory pressures reached 30 cm H2O, measured FIO2 ranged 0.65–0.95.10 Thus, as compliance or resistance worsened, patients often endured prolonged exposure to toxic levels of FIO2.
1967: ARDS, PEEP, and Oxygen Toxicity
Nineteen sixty-seven was a watershed year because of 2 studies. Ashbaugh and colleagues 11 introduced the concept of ARDS and proposed treating it with PEEP. ARDS replaced previously terms (eg, “congestive atelectasis,” “wet lung,”) that had described the phenomenon without a unifying concept. These investigators made the initial step toward understanding the underlying pathophysiology that evolved into the concept of acute lung injury.12
Equally important was the introduction of PEEP, representing the first effective therapy for treating refractory hypoxemia in ARDS and potentially avoiding O2 toxicity. Its initial use was a desperate attempt to save a young trauma patient when pressure-cycled ventilation failed. An experimental Engstrom volume ventilator with high-pressure capability was retrieved from storage. Although the expiratory retard control was unfamiliar to the authors, it was set to 10 cm H2O, resulting in rapidly improved oxygenation.13 This was largely responsible for the adoption of volume-cycled ventilation in treating ARDS requiring high ventilating pressures and V̇E demand (eg, ∼16 L/min).14
In the other study, Nash and colleagues15 described O2 toxicity as a clinical problem. However, they raised the possibility that prolonged exposure to high VT and peak airway pressures may have been contributory, thus anticipating the concept of ventilator-induced lung injury (VILI). Unfortunately, soon afterward the authors dismissed their own speculation.16 However, evidence of VILI was apparent (but unrecognized) when early ARDS investigators reflected on pre-PEEP practices. They noted radiographic opacities first appeared patchy in nature, becoming more diffuse over time and ultimately requiring “inspiratory pressures of 60–70 cm H2O” with atmospheric expiratory pressure “permit[ting] collapse of more alveoli.”14 And until the early 1990s, this was plausibly interpreted as simply representing underlying disease progression.
Missed Opportunity for Lung-Protective Ventilation
In hindsight, the reluctance to use PEEP before and after 1967 reflected the enormous influence of Cournand’s17 1947 study describing its adverse effects on cardiac output. However, that study focused not on manipulating end-expiratory pressure but on altered inspiratory:expiratory (I:E) ratios in which inadvertent PEEP was a consequence. When I:E was ≥ 1:1 during large VT ventilation, a prolonged expiratory retard (up to 2.4 s) was responsible for decreased venous return, whereas nadir end-expiratory pressure was < 5 cm H2O just before the onset of inspiration.17
Reluctance to explore PEEP prior to 1967 was curious given publications in the late 1930s on the effectiveness of continuous positive-pressure breathing in treating acute pulmonary edema.18,19 During World War II, it was also effective in treating post-traumatic wet lung syndrome.20 By 1959, PEEP was shown to increase functional residual capacity (FRC) and oxygenation during general anesthesia.21 And a 1962 symposium reflecting upon the Cournand study17 also pointed out that mean airway pressure rather than PEEP was the primary factor in hemodynamic compromise.22
A crucial opportunity was missed in not reassessing the need for large VT ventilation once PEEP was found to be effective. In addition, the singular focus on PEEP in turn biased the interpretation of other early PEEP studies. Consideration was not given to both the initial rationale for using a large VT and its singularly negative impact: the generation of both high peak intrathoracic pressures and prolonged positive-pressure decay during expiration.23 Thus, PEEP levels > 10 cm H2O were largely interpreted as dangerous and generally avoided.
Yet, in 1978 Suter and colleagues24 systematically examined the combined effects of VT and PEEP on respiratory system compliance (CRS). Increasing VT from 10–20 mL/kg caused a precipitous decline in CRS at relatively low PEEP levels (6–10 cm H2O). In contrast, when VT was limited to 5 and 7 mL/kg, CRS steadily increased even at PEEP of 15 cm H2O, thus anticipating the era of lung-protective ventilation (LPV) (Fig. 1). The curious lack of attention paid to this study likely reflected that, at the time, only 2 studies had suggested the possibility of VILI.25,26

The inter-relationship between end-expiratory pressure and tidal volume (see key) on quasi-static chest compliance resulting in patients with acute respiratory failure. From Reference 24, with permission.
In addition, ventilators of the 1970s generally lacked a direct means of measuring end-inspiratory plateau pressure (Pplat) and CRS, thus limiting the ability to assess lung stress. In order to measure Pplat, the tubing powering the expiratory “mushroom valve” had to be manually occluded to create the end-inspiratory hold. This became virtually impossible with early intermittent mandatory ventilation (IMV) requiring an external source of continuous gas flow. Thus, the impact of both PEEP and VT on lung stress was functionally removed from clinical consideration.
Both the technical limitations and the absence of consideration to pulmonary mechanics during this period were best exemplified in the super-PEEP approach to treat severe ARDS.27-29 This strategy prioritized optimizing oxygenation by targeting a PaO2 of 60–100 mm Hg on an FIO2 ≤ 0.55, a VT of 12–15 mL/kg, with reported PEEP levels of 27–54 cm H2O. In essence, oxygenation goals took precedence over the risk of lung overdistention. Pulmonary mechanics were not reported and appeared not to have factored into decision-making.
1980s: Chest Tomography and Ventilator-Induced Lung Injury Models
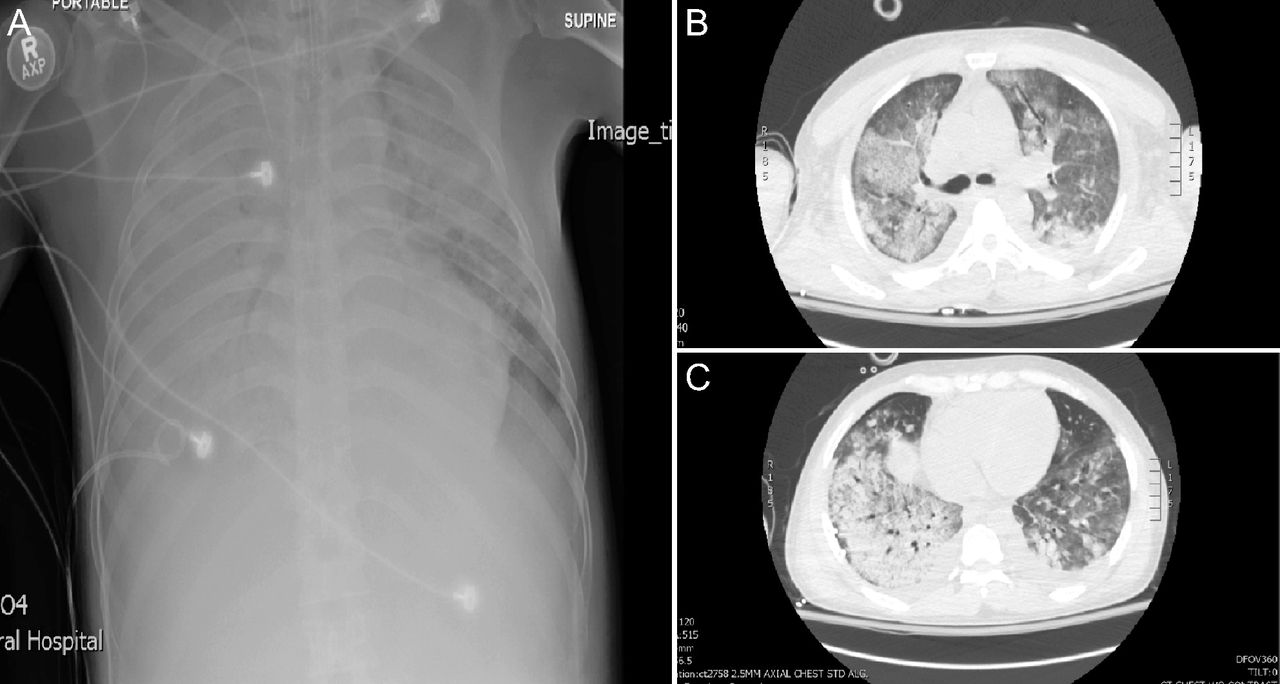
In the mid-to-late 1980s, VILI emerged as topic of concern as computed tomography (CT) studies of ARDS revealed heterogeneously distributed injury of varying intensity (Fig. 2).30,31 This led to the adult “baby lung” concept: Normal (aerated) lung tissue at end expiration in severe ARDS (200–500 g) is equivalent to that of a 5-y-old child.32 In consequence, a commonly used VT of ∼15 mL/kg functionally became ∼40 mL/kg: that used in the seminal VILI study from 1974.26 These findings buttressed other preclinical VILI studies;33-35 both reaffirming and expanding upon pioneering research done decades before.25,26 Shortly afterward came the first clinical study36 of LPV in ARDS incorporating permissive hypercapnia first used in treating status asthmaticus.37 When applied to ARDS, LPV was associated with a mortality of ∼19% (∼50% less than predicted).36

Comparison of frontal chest radiograph (A) to chest tomography images at the carinal (B) and juxta diaphragmatic (C) levels showing both the heterogenous distribution and intensity of lung injury along with areas of relatively preserved, aerated lung tissue. San Francisco General Hospital ARDS Registry, institutional review board no. 268589.
Throughout the 1990s as VILI research increased, 2 separate international consensus conferences on mechanical ventilation and ARDS strongly recommended maintaining Pplat ≤ 30 or 35 cm H2O and not > 40 cm H2O.12,38 These events coincided with the National Institutes of Health forming the ARDS Clinical Trials Network (ARDSNet) that in 2000 firmly established the efficacy of LPV in reducing ARDS-associated morbidity and mortality.39
Current Concepts in Protective Ventilation Strategies for ARDS
By the time the ARDSNet study was published in 2000, there already had been substantial progress in understanding the molecular biological mechanisms of inflammation and coagulation by which VILI develops.40 Because of this research, we came to understand the association between ARDS, VILI, and multi-organ dysfunction syndrome (MODS) that results in multi-organ failure: the primary cause of ARDS-related mortality.36,41-44 And over the past 20 years, protective ventilation has broadened from the initial 3 pillars of LPV (ie, minimizing strain, shear, and hyperoxic-related lung injury) to include right heart-protective ventilation, diaphragmatic protective ventilation, and most recently the possibility of patient self-inflicted lung injury (P-SILI).
Lung-Protective Ventilation
At its most fundamental, LPV is about matching VT to the existing FRC, which in ARDS averages 600–1,800 mL depending upon severity.45 Doing so largely prevents global and regional overdistention that causes cellular injury and exacerbates disease-associated inflammation. A VT of 4–8 mL/kg is targeted to maintain Pplat ≤ 30 cm H2O and elastic driving pressure or PDR (ie, Pplat-PEEP) ≤ 15 cm H2O. Both Pplat and PDR are indirect signifiers for peak lung stress and the change in tidal lung stress, respectively. Yet even in severe ARDS, when FRC is markedly reduced, substantial overdistention occurs in nondependent regions despite achieving targeted VT and Pplat (ie, 6 mL/kg and 28 cm H2O, respectively).46 Emerging evidence suggests targeting PDR to ≤ 13 cm H2O may reduce mortality risk further47 along with improved pulmonary function in survivors.48
Shear injury occurs when obstructed/collapsed peripheral airways and alveoli are repeatedly forced open and then re-collapse during tidal ventilation. Because heterogeneously injured lungs distribute stress unequally, most alveolar shear injury occurs at the interface between neighboring normal and injured structures. When PEEP stabilizes, newly recruited alveoli shear injury is reduced, and the impact of VT-related strain injury also might decrease by providing a larger surface area to accommodate VT without creating disproportionate stress. However, this would occur only when alveolar recruitment is greater than corresponding regional overexpansion of patent alveoli. Finally, intrapulmonary shunt and areas of low ventilation-perfusion also decrease so that FIO2 can be reduced toward nontoxic levels (ie, ≤ 0.60).9 The effects of Pplat and PEEP on recruitment and de-recruitment are discussed below.
Right Heart-Protective Ventilation
The pulmonary vasculature is a low-resistance, high-capacitance system reflected in the thin muscular wall of the right ventricle (RV), thus making it particularly vulnerable to failure with sustained elevations in pulmonary vascular resistance. ARDS is characterized by pronounced pulmonary arterial and capillary endothelial injury resulting in arterial and microvascular thrombosis,49 vascular constriction from hypoxemia, hypercapnia and acidosis,50 vascular compression from pulmonary edema,51 and alveolar hyperinflation.52 Over time, these acute changes evolve into a chronic phase characterized by vascular smooth muscle hypertrophy, fibrosis, and capillary obliteration.53
Sustained pulmonary arterial hypertension causes RV dysfunction. In ARDS, this often progresses to acute cor pulmonale (ACP), signifying right ventricular ischemia. If not reversed, it eventually causes left ventricular failure and progressive systemic hypotension.54 Prior to LPV, the incidence of ACP in ARDS often reached 60%,55 compared to 22–25% during LPV.56,57-59 In severe ARDS, however, the incidence of ACP can still reach 50%.60 Furthermore, patent foramen ovale is relatively common in severe ARDS (∼16–19%), wherein right-to-left intra-cardiac shunting further complicates ventilator management, particularly when Pplat and PEEP are highly elevated.56,61
When neither high Pplat nor high PEEP is present, ACP does not appear to increase mortality risk in ARDS.59,62 Nonetheless, severe ACP carries significantly higher mortality in ARDS than its absence (57% vs 42%, P = .03).59 Severe ACP was one of 4 factors independently associated with mortality in ARDS, the others being PaO2/FIO2 < 150 mm Hg, PDR ≥ 18 cm H2O, PaCO2 > 48 mm Hg, and pneumonia as primary etiology.59
The primary features of RV-protective ventilation are to keep Pplat < 27 cm H2O, PDR < 18 cm H2O, and PEEP ≤ 10 cm H2O. Breathing frequency is titrated to keep PaCO2 < 48 mm Hg unless intrinsic PEEP occurs (or worsens).63 If these measures are insufficient (or unfeasible), ancillary therapies such as prone position, extracorporeal membrane oxygenation (ECMO) or extracorporeal CO2 removal, and inhaled vasodilators are recommended.63 Improved RV function and/or reduced pulmonary arterial pressure in ARDS also occurs with prone position60 and inhaled vasodilators.64-67 In addition, the largest ECMO trial in ARDS reported substantial reductions in Pplat, PEEP, and PaCO2 with increased oxygenation, suggesting corresponding improvements in RV function68 that previously had been observed in case reports.69
Power Transfer in VILI and the Role of Extracorporeal Support
During inspiration, the lungs store energy needed to overcome tissue elastic recoil that in turn drives passive exhalation. This fact has modified the concept of VILI and LPV to propose that V̇E demand plays a role because the repetitiveness of applied strain/stress represents power transferred from the ventilator to the lungs. This, however, involves 3 specific preconditions (caveats): (1) the relative size of the baby lung (ARDS severity) that determines tissue capacity to receive tidal energy pulses, (2) the relative durability (or fragility) of injured and non-injured tissue to endure repetitive energy pulses, and (3) the duration or cumulative power transfer following VILI onset.70,71 Mechanical power transfer to the lungs > 12 J/min is associated with VILI in animal models72 and has been independently associated with mortality in ARDS.47
Elevated V̇E demand in ARDS reflects both metabolic rate and physiologic dead-space fraction that increases markedly from mild to severe ARDS, whereas CRS correspondingly decreases.73 Thus, the highest energy transfer potentially exacerbating VILI occurs in severe ARDS when lung injury (and tissue fragility) is most acute. Although power transfer can be lessened by inducing permissive hypercapnia, this too has inherent drawbacks. Hypercapnia-induced increases in respiratory drive evokes severe dyspnea and likely potentiates asynchrony,74 the suppression of which requires generous use of sedatives and often the addition of neuromuscular blockade.
In this context, either ECMO or extracorporeal CO2 removal has substantial merit. Lung protection afforded by venovenous ECMO in ARDS primarily has focused upon oxygenating venous blood to reduce VILI risk by reducing FIO2 and PEEP requirements. In contrast, venoarterial ECMO partially supports systemic O2 delivery when cardiac output is impaired. ECMO has been used in severe ARDS since the early 1970s.75 However, because contemporaneous practices included large VT ventilation, the potential benefits of ECMO were not realized (mortality ≥ 90% with or without ECMO).76
By 1984, extracorporeal support in ARDS had incorporated the idea of combining extracorporeal CO2 removal with low-frequency pressure controlled inverse-ratio ventilation (PC-IRV), limiting Pplat to 30–35 cm H2O to promote lung rest.77 A small observational study78 reported a surprisingly low mortality of 23% (projected mortality > 90%) that was difficult to interpret given numerous methodological issues. In 1994, a randomized controlled trial of “lung-rest” that combined PC-IRV (Pplat/PEEP: 45/24 cm H2O, VT: 3 mL/kg, rate: 3) with extracorporeal CO2 removal was compared to protocolized volume ventilation with high PEEP (16 cm H2O) and moderate VT (10 mL/kg). Mortality was not different (67% vs 58%, respectively, P = .80). The 2009 CESAR trial compared a rest strategy consisting of pressure controlled ventilation (Pplat/PEEP: 20–25/10–15 cm H2O, rate: 10, FIO2: 0.30) with venovenous ECMO to the ARDSNet lower-PEEP LPV strategy.79 Mortality was substantially lower in the ECMO arm (37% vs 49%, respectively). However, among other methodological issues, there was no documentation of actual adherence to the ARDSNet protocol (ie, use of the protocol was only encouraged). This raised serious concerns over the study’s internal validity.80
The most recent randomized controlled trials of ECMO68 and extracorporeal CO2 removal 81 for ARDS also found no mortality benefit compared to well-established protocolized LPV management used in the EXPRESS82 and ARDSNet39 trials. However, the results of the EOLIA trial68 were intriguing given the clear trend toward improved mortality favoring ECMO (35% vs 46%, respectively, P = .09) and significant improvement when analyzed according to treatment failure (ie, mortality in the ECMO arm vs mortality and crossover to ECMO therapy in the LPV arm) (35% vs 58%, respectively, P < .001).
However, the crossover group had more severe lung injury at randomization and higher mortality compared to control arm subjects not requiring ECMO (57% vs 41%, respectively).83 This intention-to-treat trial was terminated early for futility, leaving the study’s interpretation ambiguous. One plausible interpretation is that the higher mortality in the control arm was due to the sicker crossover subjects who did not appear to benefit from ECMO. Both treatment arms made very liberal use of the full array adjunctive therapies used in severe ARDS. Hence, continued deterioration in severe ARDS despite LPV with higher PEEP and multiple ancillary therapies suggests that the addition of ECMO is unlikely to be beneficial.
ECMO as rescue therapy in severe ARDS will likely continue. Slow enrollment in the EOLIA trial (which took 6 years) suggests that ECMO is reasonably applicable only to a small fraction of ARDS cases. Furthermore, the clearly negative results of the REST trial81 and high incidence of serious adverse events (31%) render routine use of extracorporeal CO2 removal in less severe ARDS unlikely.
Respiratory Muscle Physiology and Mechanical Ventilation
Respiratory muscle physiology research greatly informed our understanding of patient-ventilator interactions, ventilator dependence, and the potential exacerbation of both VILI and MODS. Beginning in the mid-1970s, loaded breathing and muscle fatigue became a focus of interest,84-86 with studies relevant to assisted mechanical ventilation emerging a decade later.87-91 These studies demonstrated that patient effort continues throughout most or all of mechanical inspiration. Prior to this, there had been a pervasive lack of curiosity despite decades’ long clinical encounters of patients “fighting the ventilator.”3 These studies flipped the narrative to the ventilator (and thus the clinician) “fighting the patient.”
Diaphragmatic Protective Ventilation
That work of breathing often exceeded physiologic resting levels raised particular concern that patients with diaphragmatic fatigue/failure might receive inadequate support necessary to facilitate recovery. This prompted additional exploration into respiratory muscle injury during critical illness that distinguished 2 opposing mechanisms. Use atrophy describes respiratory muscle inflammation from exposure to excessive workloads,92 both in healthy individuals and those with chronic lung disease. This occurs even following brief exposure to maximal loading.93 Clinically, the phenomenon of delayed diaphragmatic injury and inflammation (occurring 3 d after brief periods of intensive loading) produced diaphragmatic weakness,94,95 rendering the diaphragm more susceptible to further fatigue and injury. Thus, disallowing a period of full rest following either acute respiratory failure onset or after a failed weaning trial might induce chronic fatigue and, paradoxically, prolong ventilator dependence.
In addition, sepsis causes diaphragmatic injury, wherein the diaphragm exhibits exaggerated pro-inflammatory gene expression and hence cytokine production.96 Passive mechanical ventilation in sepsis substantially reduced muscle injury and improved diaphragmatic force generation.97 At ICU admission, the majority (64%) of subjects with acute respiratory failure exhibited diaphragmatic weakness associated with either sepsis or disease severity that carries a poorer prognosis.98 Thus, attributing diaphragmatic dysfunction primarily to mechanical ventilation practice can be misleading.
In contrast, ventilator-induced diaphragmatic dysfunction (or disuse atrophy) is the progressive loss of diaphragmatic force-generating capacity (eg, ∼40–50%) from prolonged periods of passive ventilation and loss of electromyographic stimulation, the duration of which appears species-dependent (eg, 1 d in rabbits vs 11 d in baboons).99 In critically ill subjects undergoing prolonged passive ventilation, diaphragmatic strength progressively decreased by 32 ± 6% over 6 d (becoming prominent at ∼3–4 d).100 These findings coincided with evidence of muscle fiber injury and muscle atrophy. Resumption of muscular contraction following disuse atrophy likely increases vulnerability to load-induced injury.99
Conversely, in animal models of spontaneously triggered mechanical ventilation, diaphragmatic weakness is ameliorated substantially compared to passive ventilation with strength reductions of 14% versus 48%, respectively, over 3 d.101 Other studies found periodic interruptions of passive ventilation with spontaneous breathing attenuated diaphragmatic mass and strength loss compared to passive ventilation. In a clinical study in which biopsies were obtained, subjects capable of generating spontaneous efforts ∼36% of the time over an 8-d course had significantly less diaphragmatic injury compared to those with a 3-d course of passive ventilation.102
In summary, the strategy of diaphragmatic protective ventilation involves a 2-pronged approach involving both sedation and ventilator settings.103 First is to limit the duration and incidences of passive ventilation and prevent excessive ventilatory support that needlessly suppresses patient effort. Second is avoiding prolonged periods of highly loaded breathing, particularly early in the course of acute respiratory failure. Many of these patients present with acute diaphragmatic weakness, muscle injury, and fatigue, the resolution of which (depending upon several factors) may require from one to several days.88,98,104
The Concept of Patient Self-Induced Lung Injury in ARDS
Lung stress is trans-alveolar pressure generated either by positive airway pressure or negative pleural pressure from muscular contraction and is the basis of the push-pull concept of assisted breathing.105 Under normal lung physiology, distending pressure across fluid-like structures transmits stress relatively evenly such that the risk of VILI or P-SILI is likely minimized except under extreme conditions (see below).106
By the late 1980s, studies on healthy animals in whom hyperventilation was either chemically induced (rate: 69, VT: 9 mL/kg),107 or by negative-pressure mechanical ventilation (rate: 25, VT: 44 mL/kg),33 produced acute lung injury within a matter of minutes or hours (depending upon VT size). Histological examination found diffuse alveolar damage and altered permeability edema.33
Hydrostatic mechanisms also may have contributed because intense negative intrathoracic pressure increases both venous return and left ventricular afterload, leading to pulmonary engorgement and pulmonary hypertension.108 A similar mechanism appears responsible for transient acute lung injury in elite athletes due to pulmonary capillary stress failure.109 When comparing positive- versus negative-pressure ventilation, one study found substantially greater pulmonary edema generated by negative intrapleural pressure versus positive pulmonary intravascular pressure despite the same VT and relative driving pressure.33
The significance of initial preclinical research was not appreciated until clinical studies of early neuromuscular blockade in ARDS reported both significantly reduced mortality and mechanical ventilation duration.110 A related study found significantly reduced pro-inflammatory mediators both in pulmonary edema fluid and serum.111 Thus, combining evidence that PDR ≤ 15 cm H2O reduces mortality risk in ARDS,112 with aforementioned preclinical evidence, and clinical studies showing large esophageal pressure swings commonly occur during assisted ventilation in ARDS raised the possibility that vigorous spontaneous breathing efforts may exacerbate lung injury and worsen outcomes.113,114 Concern over P-SILI is supported by anecdotal evidence of extreme esophageal pressures swings of 35 cm H2O during LPV in severe ARDS,115,116 one study observing extraordinarily severe alveolar edema.115
Mean esophageal pressure swings of 14–17 cm H2O have been observed in sedated subjects with ARDS during assisted LPV at ∼7 mL/kg VT.117 Preclinical acute lung injury models of LPV (VT: ∼8 mL/kg, Pplat: 30 cm H2O) found inspiratory esophageal pressures of only 5 cm H2O produced transpulmonary pressures of 35 cm H2O that exacerbated lung injury.118
However, the negative impact of spontaneous breathing may depend upon the underlying severity of acute lung injury.119 In the aforementioned preclinical model, spontaneous breathing during mild lung injury occurred at both a lower Pplat and lower effort, causing dorsal lung recruitment with improved oxygenation and little histological impact (compared to passive ventilation). In contrast, severe lung injury stimulated greater spontaneous effort at a higher baseline Pplat that (paradoxically) promoted cyclical de-recruitment, worsening both oxygenation and lung injury compared to passive ventilation (see below). Preclinical models also observed occult pendelluft motion wherein diaphragmatic contraction redistributes alveolar volume away from nondependent to dependent lung regions, causing paradoxical overdistention of dependent lung without changing delivered VT.120
Another aspect of P-SILI is that excessive breathing efforts in the acute phase of ARDS foment fluid transudation, worsening pulmonary edema, gas exchange, and CRS.115 This is further exacerbated by compensatory expiratory muscle recruitment during loaded breathing that promotes de-recruitment by creating a counterforce to applied PEEP.121 In contrast, during passive ventilation, PEEP facilitates pulmonary edema clearance122,123 that in ARDS already is markedly depressed.124 Moreover, the inability to clear pulmonary edema in ARDS is associated with increased mortality.125
It’s important to recognize we are in the very early stages of understanding the potential clinical importance of P-SILI. The extent of lung injury induced by negative pleural pressure swings when the resulting global strain is limited to 5–8 mL/kg versus 12–15 mL/kg is likely substantial and clinically relevant in terms of balancing risk factors affecting outcomes. These issues must be better understood before altering sedation practices as prophylaxis against P-SILI (see below).
The Implications of Spontaneous Breathing During Critical Illness
Prior to the advent of IMV, all weaning was done by spontaneous breathing trials (SBTs), reflecting the limitations of ventilator technology.126 The advantages afforded by IMV during this period were (1) avoidance of hypocarbia during assisted ventilation with elevated respiratory drive, (2) countering cardiac output depression associated with high PEEP, and (3) providing partial V̇E support in difficult-to-wean patients by allowing longer periods of exercise reconditioning without excessive discomfort.127
IMV emerged partly as a byproduct of critical care practices that successfully stabilized patients with catastrophic illness or injury that in turn promoted survival to the recovery phase, albeit often in a severely debilitated state. This preceded our awareness of critical illness-related myopathies and neuropathies128 as well as providing appropriate nutritional support.129 Thus, clinicians increasingly encountered some patients who were difficult to wean for which IMV provided a reasonable solution.130
What began as a recovery phase strategy transformed into a popular primary ventilation mode. At the time, the rationale supporting IMV was met with criticism127,131 along with evidence that it did not improve weaning efficacy.132,133 Later on, it was demonstrated that as IMV rate decreased respiratory drive increased so that mandatory breaths did not reduce patient work of breathing (Fig. 3).91 This likely increased the risk for acute diaphragmatic injury as well as chronic diaphragmatic fatigue.

Differences in patient work of breathing between patient-triggered manual breaths during synchronized intermittent mandatory ventilation (SIMV triggered) and corresponding unsupported spontaneous breathing as the mandatory rate is reduced from 100% (ie, all efforts resulting in a triggered mandatory breath) to 0% (ie, breathing on continuous positive airway pressure through the ventilator). Note that as mandatory breaths are reduced the resulting increase in respiratory drive and work of breathing performed during spontaneous efforts continues during mandatory breaths as well, thus reducing the effectiveness of mandatory breaths to offload the inspiratory muscles. Data from Reference 91.
Although early SBT attempts in debilitated patients were poorly tolerated, the unappreciated advantage was that clinicians could intervene quickly to provide appropriate periods of diaphragmatic rest and rapid recovery (ie, avoidance of chronic diaphragmatic fatigue).134-136 In contrast, IMV proponents recommended progressively reducing the mandatory rate “as long as spontaneous respiratory efforts occurred and arterial pH remained > 7.30 regardless of the other measurements” (emphasis added).137
The rationale for IMV as a primary mode of ventilation was based upon misperceptions involving both of the frequency of difficult weaning and the nature of its origins.135,138 What followed was a positive feedback loop: As IMV gained popularity so too did the illusion that most patients were likely difficult to wean, requiring a gentle transition to unassisted breathing.131 The philosophy of gradual weaning seamlessly transferred over to pressure support ventilation (PSV) in the 1980s. The tendency to always initiate PSV at elevated levels was dubiously interpreted as actively weaning.
Gradual weaning as standard practice was discredited by large prospective trials in the mid-to-late 1990s. Regardless of weaning modality, the strategy of gradual withdrawal actually increased mechanical ventilation duration by 6–9 d.139 In addition, a once daily SBT was found superior to both IMV and PSV in reducing weaning duration.140 Most telling, a major study examining weaning techniques in acute respiratory failure found 89% of potentially eligible patients passed their initial screening SBT and, therefore, could not be enrolled.140 This affirmed previous observations that more than 70% of mechanically ventilated patients resume unassisted breathing without difficulty.138 In addition, protocolized daily screening for weaning readiness followed by an SBT significantly reduced mechanical ventilation duration.141 In ARDS, the combination of daily screening, SBT, and conservative sedation practices reduced both median duration of mechanical ventilation and ICU length of stay by 5 d.142
Recruitment and De-recruitment in ARDS
The Legacy of “Congestive Atelectasis”
“Contrary to widespread belief, true anatomic atelectasis is not greatly contributory to the observed pathophysiology and response to treatment; specifically the severity, duration and potential reversibility of increased pulmonary capillary endothelial permeability and the resultant magnitude and persistence of transcapillary leakage of both fluid and particularly serum protein are major determinants for a wide variation in clinical course”(emphasis added).
Carl Teplitz143
A lingering problem in understanding how best to ventilate patients with ARDS was inherited from the 1950s. Congestive atelectasis was coined by Jenkins and colleagues144 to describe pathologic findings from postoperative respiratory failure that became commonplace, particularly in trauma patients during both World War II20 and the Vietnam war.145,146 However, the original case series by Jenkins et al consisted mostly of subjects with abdominal infection or trauma who developed hemodynamic instability and fever, suggesting septic shock.144 The primary pathology findings were pronounced capillary congestion and alveolar hemorrhage, with atelectasis being a prominent finding only in a minority of subjects.144
The term congestive atelectasis also was used by Ashbaugh and colleagues in describing ARDS, but their vague wording suggested atelectasis, interstitial-alveolar hemorrhage, and pulmonary edema were equally prevalent findings.11 In contrast, both contemporaneous15,147 and subsequent pathologic studies found alveolar filling (not atelectasis) was the primary abnormality during the early exudative phase of ARDS.143,148-152 After 1967, the modifier congestive was dropped, whereas curiously the descriptor atelectasis persisted.
This had an unintended consequence wherein atelectasis has been misinterpreted to imply acutely injured lungs can potentially be fully recruited and thus (at least subliminally) implying a return to a normal state. This in turn facilitated the idea that patient outcomes might improve with open lung-oriented strategies favoring higher PEEP and recruitment maneuvers, PC-IRV, airway pressure release ventilation (APRV), and high-frequency oscillatory ventilation (HFOV). Yet, when eventually tested in large randomized controlled trials (if ever seriously attempted), the results have been disappointing.153-155
Although recruitment and de-recruitment were implicitly discussed since the beginning of ICU mechanical ventilation,7,14,156 it wasn’t until the early 1990s with the advent of open lung ventilation157 that the discussion became explicit and a focal point of attention. The idea of alveolar recruitment and de-recruitment in ARDS was met with skepticism and has highlighted the ambiguous nature of what actually occurs when we attempt to open up the lungs. In particular, the overly liberal usage of atelectasis when alveolar flooding is the primary lesion has been problematic.158 With certain exceptions (eg, obstructive lobar collapse, abdominal compartment syndrome), profound tidal collapse in ARDS is unlikely. Moreover, equating de-recruitment with nonaerated tissue is also problematic because its usage confuses fundamentally different lesions: degassed/collapsed alveoli versus alveolar filling.158
In oleic acid models of acute lung injury (which closely mimics the exudative phase of ARDS),159 decreased gas volume in dependent zones does not cause collapse because of alveolar filling.160,161 This created considerable problems interpreting the lower inflection point in pressure-volume curves. At first believed to signify full alveolar recruitment, it was later interpreted as the beginning of recruitment in the nondependent injured lung.162 Yet, a lower inflection point may represent the initial impedance to inflation of an edematous, non-collapsed lung; the transition from liquid-filled to gas-filled airspaces by displacing edema158; or overcoming intrinsic PEEP.162
This is not to suggest compressive atelectasis plays no role in ARDS. Rather, it is to re-emphasize and redirect our attention to the key role played by peripheral airway obstruction/collapse and alveolar filling in ARDS. Most importantly, full lung recruitment in ARDS is neither plausible nor necessary. CT studies examining lung recruitment (whatever that actually signifies) estimated that tissue consolidation accounts for ∼25% of the lung in ARDS.163 Moreover, improved oxygenation in ARDS has not translated into clinically meaningful outcomes.39,153 Mortality in ARDS is caused primarily by the presence or development of MODS, which is intimately associated with VILI and not severe refractory hypoxemia (see below).41,42,164,165
Infant Respiratory Distress Syndrome and Open Lung Ventilation
The theoretical basis for open lung ventilation (upon which HFOV, PC-IRV, and APRV are conceptually associated) originated in treating infant respiratory distress syndrome.166,167 Studying the effects of mechanical ventilation in hyaline membrane disease used non-injurious surfactant washout models that largely produce homogenous lung collapse as well as homogenous recruitment/reinflation.168-170 These models deviated fundamentally from the mechanics and pathology of ARDS and, therefore, tended to facilitate misleading generalizations about the potential effectiveness of open lung strategies for treating ARDS. It is not coincidental that prior to introducing the concept of open lung ventilation in 1991157 much of Dr Lachman’s prior work focused on the treatment of hyaline membrane disease.171-173
Fast Versus Slow Pulmonary Compartments
An underappreciated aspect of ARDS is the presence of fast and slow pulmonary compartments reflecting the distribution and severity of lung injury. This has shaped our perceptions of recruitment potential as well as de-recruitment; that in turn influenced how we interpreted the effectiveness of recruitment at any setting of Pplat and PEEP.174 Furthermore, historically we assessed the effectiveness of PEEP on oxygenation between 10–15 min.175 Perhaps valid during the fibrotic end stage of ARDS,176 it is highly questionable in the exudative phase of moderate to severe ARDS.
The presence of slow compartments has been observed during PC-IRV in infants,167 in response to PEEP,176-179 PC-IRV in adults,180 and prone positioning.181 Prospective studies that carefully examined the presence and time course of pulmonary compartments in ARDS observed time periods ranging from several minutes to several hours.177,178,182
What was being described likely reflected the effects of reopening collapsed or obstructed peripheral airways/alveolar ducts and redistribution of pulmonary edema, the time course of which is determined by several factors (eg, the amount and viscosity of edema, airway film surface tension, radial traction supporting peripheral airways, physical obstruction by fluid plugs, presence and amount of functional surfactant, and the presence of inflammatory cells and cellular debris).174 These in turn determine how the interplay of applied airway pressure and maneuver duration impacts the potential effectiveness of recruitment maneuvers and recruitment enhancing modes. Furthermore, the time course for recruitment within a single breath is very limited (ie, ∼85% occurring within 2 s).183
Two clinical implications issue from this. First, radical time inversions during PC-IRV or APRV (eg, 2:1 and beyond) have little effectiveness in terms of recruitment. More importantly, oxygenation improvements in these approaches likely reflect the effects of intrinsic PEEP, the distribution of which is uncertain and perhaps higher in less injured or normal tissue.184 In contrast, the negative consequence of sustained high intrathoracic pressure includes perpetuating strain injury in nondependent regions, increased risk of cor pulmonale, and compromised hemodynamics,160 thus increasing the risk of hypoperfusion and ischemia to vital exchange organs that may perpetuate systemic inflammation (see below).
An initial rationale for inverse-ratio ventilation in ARDS was the hypothesis that as expiration proceeds underinflated alveoli become increasingly susceptible to hydrostatic compressive forces. Therefore, shortening expiratory time in order “to catch the lung at a critical volume on its way down” would reduce the risk of collapse.185,186 However, de-recruitment in dependent lung regions generally begins at PEEP < 15 cm H2O and infrequently < 20 cm H2O,174 thus obviating the need for time inversion in managing ARDS.
Second, the observed response of the slow pulmonary compartment, as evidenced by progressive recruitment over time, suggests that recruitment of obstructed and collapsed peripheral airspaces requires repetitive application of pressure during tidal ventilation over an extended time period. And because recruitment maneuvers are applied for only a few minutes, the actual or optimal efficacy of any tested pressure levels remains unknown.
Finally, the theoretical attractiveness of HFOV was that it prevented VILI by avoiding convective gas flow and the effects of regional time-constant differences in causing regional lung overdistention.187 But these very problems likely occurred during HFOV because regional time-constant abnormalities probably resulted in regional static overinflation.170,188,189 This was apparent in one trial by both higher vasopressor requirements and incidences of barotrauma.155
Current Understanding and Implications of Recruitment and De-recruitment in ARDS
CT studies observed that maximal recruitment in the middle lung regions occurs between a Pplat of 20–30 cm H2O, whereas dorsal zones begin to recruit at a Pplat of ∼20 cm H2O, with the largest incremental change in dorsal aeration also occurring at a Pplat of 30 cm H2O.190,191 In ARDS, de-recruitment is a continuous process that usually becomes prominent at PEEP < 15 cm H2O191 and is both pronounced and rapid dorsally when PEEP falls below 10 cm H2O.191,192,193
These findings imply that in the absence of pronounced extrathoracic compressive forces PEEP levels of ∼10–15 cm H2O at a VT producing a Pplat of 25−30 cm H2O are likely sufficient to achieve adequate oxygenation at a relatively nontoxic FIO2. This has been repeatedly confirmed in several major LPV studies (Table 1).39,82,194,195
Lung-Protective Ventilation Characteristics Over the First 3 Study Days in Major Trials
PEEP: Curative Versus Supportive Therapy
An intriguing aspect of recruitment-oriented ventilation strategies in ARDS (ie, IMV/super PEEP, PC-IRV, and APRV) has been its advocacy in trauma care.29,196,197 Trauma-associated ARDS often involves large volume resuscitation with fluids and blood products that augment lung injury through indirect pathways (ie, transfusion-related acute lung injury or TRALI).198,199 Trauma-induced capillary leak syndrome (TICS) commonly results from severe injury exacerbated by crystalloid solutions in a condition known as transfusion-associated circulatory overload. The persistence of TICS following injury is complex and variable,200 whereas the clinical presentation of transfusion-associated circulatory overload often overlaps with and mimics TRALI.198
TICS may cause further complications (ie, anasarca, ascites, and abdominal compartment syndrome) that often require recruitment-oriented ventilation strategies. PEEP counters intraparenchymal hydrostatic forces, that promote pulmonary edema formation and compressive forces associated with decreased chest wall compliance.
In addition, a prominent feature of TICS is severe hypoproteinemia.200 In ARDS, alveolar edema has a protein concentration similar to plasma (ie, ≥ 75%)201 so that TICS-induced hypoproteinemia lowers edema viscosity, thereby reducing the yield pressure and time needed to recruit obstructed peripheral airways and redistribute edema from the alveolar space.174 This likely enhances the efficacy of recruitment to a greater extent than what otherwise occurs in ARDS without overhydration (eg, pneumonia). Ashbaugh and colleagues suggested as much observing that PEEP appeared to reverse the syndrome in fluid-overloaded trauma subjects.11 This gave rise to the illusion of PEEP as curative. In contrast, Ashbaugh and colleagues observed that in ARDS associated with pneumonia or aspiration “the illness was likely to be prolonged and refractory to treatment.”14
The misconception that PEEP is curative was long embraced by IMV/super-PEEP proponents, even suggesting PEEP or CPAP might prevent ARDS.202 The illusion of PEEP as curative reflected the fact that early onset (eg, ∼4 d) trauma-associated ARDS is associated with lower mortality among patients who generally are younger and healthier than other ARDS etiologies.164,203-205 Moreover, trauma-induced ARDS is associated with significantly less endothelial and epithelial injury, supporting the impression that it may be self-limiting in nature.203,205 The pro-curative PEEP argument preceded our understanding of systemic inflammatory response syndrome, wherein ARDS often represents “the pulmonary manifestation of multiple failing organs.”206
VILI, Organ Cross Talk, ARDS Phenotypes, and Multi-Organ Dysfunction Syndrome
VILI and diaphragmatic injury contribute to systemic inflammation leading to MODS and eventually multi-organ failure: the primary determinant of ARDS mortality.41-44,164,165,207 This occurs via organ cross talk by which capillary endothelial injury in one organ (generating pro-inflammatory, procoagulant, chemoattractant mediators) spills over into the systemic circulation activating an inflammatory response in the endothelium of distant organs.165,206,208,209 Highly vascularized exchange organs (eg, kidneys, intestines, liver) tend to be the initial and most vulnerable organs, in turn instigating a self-perpetuating injury pattern. In ARDS, pulmonary-renal dysfunction is an early manifestation of MODS,210 with profound consequences as secondary renal failure in ARDS increases mortality (ie, ∼60−80%).208
Despite decades of research, most therapies used in ARDS (particularly pharmacologic) have been ineffective. Thus, it has become increasingly apparent that clinical criteria used to define syndromes such as ARDS and sepsis are inherently inadequate in understanding the underlying biology necessary for developing effective drug treatments as well as more refined approaches to mechanical ventilation.211,212 Particular to ARDS is the variety of injurious agents fomenting acute lung injury, the wide range of physiologic abnormalities it produces, the related biological pathways and gene expression involved, and how the syndrome evolves or resolves over time.
Current trends in ARDS research involve devising strategies based upon recent advances in our understanding. This includes varied clinical presentations such as lobar versus diffuse lung injury, severity of hypoxemia, and most recently the interactions between pro-inflammatory mediators with markers of endothelial dysfunction that distinguish early resolution from persistent hypoxemia.213 Newer still is examining specific gene expression governing inflammation that might provide a more comprehensive understanding of ARDS, and also the possibility for highly specific novel drug therapies for certain ARDS phenotypes, or perhaps targeting current therapies more effectively. Research in this area already has discerned ARDS sub-phenotypes associated with the spectrum of inflammatory response (ie, uninflamed, reactive, hyporeactive, and hyperreactive), suggesting the existence of treatable traits with implications beyond ARDS.214 The hoped-for result is developing more personalized medicine for the treatment of ARDS and other critical illnesses.
“Wither Goest Thou?”: Integration of Ventilator and ICU Monitoring Data With Other Health Systems Data, ARDS Phenotypes, Computational Intelligence, and Closed-Loop Ventilation
The 60-year history of ICU mechanical ventilation witnessed enormous growth in our understanding of ARDS and critical illness generally as well as breathtaking advances in technology. These have vastly improved the quality of care delivered compared to that received in 1961. However, misperceptions, misconceptions, and missed opportunities occurred along the way that negatively impacted patient outcomes, some of which have taken generations to overcome or still persist. To some degree this was inevitable as our comprehension of ARDS came from integrating accrued knowledge in a process akin to creating a mosaic: The image cannot be recognized until a sufficient number of pieces are identified and placed in proper relationship to one another.
Unfortunately, that only represents part of the problem. The other problem has been an all-too-human propensity to resist abandoning long-held practices that are no longer relevant. All ventilator modes and associated practices were created during a specific historical period, reflecting the limitations of our understanding at that moment (eg, IMV, PC-IRV, APRV). The broader implications of this reluctance were recently commented on that “despite strong evidence and explicit guidelines for delivering state of the art care, adoption of current best practices for using LPV to treat ARDS have lagged.”215
Resistance to change, I believe, is partly explained by certain innate perceptual limitations. An example is the 22% relative mortality reduction found in the ARDSNet low VT study.39 This finding projected a number to treat whereby for every 11 ARDS patients treated with a 6 mL/kg VT one additional life is saved. Hence, the reality that injurious ventilation increased mortality was too subtle to be perceived in clinical practice. The legacy of our ∼200,000-year-old nervous system was its primary orientation toward interpreting real-time phenomena that promoted our survival in a radically different environment.216 Therefore, our ability to fully comprehend complex phenomena such as ARDS mortality can only be discovered by the latent potential our nervous system afforded us: the capacity for highly abstract thought and computational skills. However, the complex reality of ARDS is too often obscured by the potency and immediacy of our impressions formed in an emotionally-charged environment that is the ICU. And this brings us to the threshold of “whither goest thou?”
Further progress toward improved outcomes in ARDS will require machine learning (artificial neural networks) for understanding biologically discrete clusters of phenotypes using multiple protein biomarkers that might lead to highly targeted therapies.212 Moreover, this potential will expand considerably with the integration of ARDS genomics. The transcriptomics of nucleic acid-based arrays has already broadened the scope of exploration to tens of thousands of genes, providing a more comprehensive overview for understanding the pathophysiology ARDS and the potential for creating personalized medicine.212
And whereas molecular biology and genomics suggest potential breakthroughs in the decades to come, near-term advances in care might be closer. We can now collect massive amounts of moment-by-moment data from ventilators, monitors, and other devices in the ICU that can be filtered, uploaded, and integrated with much larger databases containing laboratory results, demographics, comorbidities, and other information to create much grander mosaics of what occurs dynamically during the course of critical illnesses.
Such computational intelligence generated by artificial neural networks performs tasks such as pattern association and pattern recognition including algorithms oriented to biostatistical multivariable analyses.217 Discovered patterns then can predict future data (eg, model development and testing) or perform other kinds of decision making under uncertainty (eg, perhaps closed-loop ventilation). One such method (ie, radial basis functions) was developed over 40 years ago by visionaries who created computerized geometric displays of cardiorespiratory and metabolic variables. These were plotted as a wheel and graded according to mean ± 4 SDs using normalized control subjects.218,219 This computerized schema was used at first to identify and then to chart the movement of individual patients over time and their response to discrete therapeutic interventions in the ICU (Fig. 4).

An early example of computational intelligence using radial basis functions to plot cardiorespiratory variables in a surgical patient. The center plot representing the physiologic response to an unspecified therapy. The 4 surrounding reference plots represent archetypal patterns expressing different physiologic states. From upper left moving clockwise: A state represents a balanced or healthy physiologic response to surgical stress, whereas states B through D represent an increasingly unbalanced pathological trajectory toward the development of multi-organ failure (MOF) and a pre-moribund state. The computer program could calculate the characteristics of any current state and the relative distance and movement toward or away from other states over time and/or in response to interventions. From Reference 218.
Given current and future advances in supercomputing, similar schemas applied to much broader data capture might be used to study ARDS, sepsis, and other critical illnesses to better understand pathophysiology dynamically and its response to contemporary therapies. This could be accomplished on a time scale far beyond our cognitive capacity. And once integrated with biomarkers and genomics, these findings might shed light on potential therapies tailored and timed to specific ARDS phenotypes that then could be assessed for efficacy.
Scaled-down versions of similar temporal plots potentially could be developed for ventilator management and integrated with other monitoring devices. In consequence, it may be possible for computational intelligence to assess and assist (eg, through advisories) ventilator management or perhaps direct closed-loop interventions in response to numerous other inputs (eg, blood pressure, stress index, volumetric capnography). The possibilities for how mechanical ventilation might be adjusted in treating ARDS 20 years from now is as unimaginable today as it was for us 50 years ago to imagine current practices when the Bennett MA-1 ventilator represented the forefront of technology.
The practice of performing hand-gathered ventilator and gas exchange data and entering it into a computer for snapshot status assessments will soon vanish (at least in countries with advanced health care delivery). It is unlikely that we will see new ventilator modes and more likely that automated adjustments based on more sophisticated monitoring will utilize existing volume, pressure, or dual-control mode technology.
Respiratory therapists are (and I believe will continue to be) indispensable members of the critical care team. That was very apparent during the first technological boom of the mid-1980s, when both ventilator modes and associated monitoring achieved a level of sophistication exceeding the practical limitations (or inclinations) of other critical care practitioners.220 However, our cognitive skill set will need to keep pace in order to remain relevant. Most importantly, as our understanding of ARDS and the role of mechanical ventilation advances, we need to avoid the all-too-human tendency to hold on to beliefs instilled in us at earlier points in our career. Such attitudes are counterproductive to improving patient outcomes. And so, it is only fitting to give the last word to our colleague, mentor, and dear friend Bob Kacmarek, who many years ago famously challenged our profession with the command Carpe Diem!221
Footnotes
- Correspondence: Richard H Kallet MSc RRT FAARC, 2070 Fell St. Apt 1, San Francisco, California 94117. E-mail: richkallet{at}gmail.com
Mr Kallet discloses relationships with Asthma and Allergy Prevention Company, ContinuED, and Lungpacer Medical.
A version of this paper was presented by Mr Kallet as the first Robert M Kacmarek Scientific Memorial Lecture at AARC Congress 2021 LIVE!, held virtually, on December 7, 2021.
- Copyright © 2022 by Daedalus Enterprises
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.
- 66.
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.
- 86.↵
- 87.↵
- 88.↵
- 89.
- 90.
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 113.↵
- 114.↵
- 115.↵
- 116.↵
- 117.↵
- 118.↵
- 119.↵
- 120.↵
- 121.↵
- 122.↵
- 123.↵
- 124.↵
- 125.↵
- 126.↵
- 127.↵
- 128.↵
- 129.↵
- 130.↵
- 131.↵
- 132.↵
- 133.↵
- 134.↵
- 135.↵
- 136.↵
- 137.↵
- 138.↵
- 139.↵
- 140.↵
- 141.↵
- 142.↵
- 143.↵
- 144.↵
- 145.↵
- 146.↵
- 147.↵
- 148.↵
- 149.
- 150.
- 151.
- 152.↵
- 153.↵
- 154.
- 155.↵
- 156.↵
- 157.↵
- 158.↵
- 159.↵
- 160.↵
- 161.↵
- 162.↵
- 163.↵
- 164.↵
- 165.↵
- 166.↵
- 167.↵
- 168.↵
- 169.
- 170.↵
- 171.↵
- 172.
- 173.↵
- 174.↵
- 175.↵
- 176.↵
- 177.↵
- 178.↵
- 179.↵
- 180.↵
- 181.↵
- 182.↵
- 183.↵
- 184.↵
- 185.↵
- 186.↵
- 187.↵
- 188.↵
- 189.↵
- 190.↵
- 191.↵
- 192.↵
- 193.↵
- 194.↵
- 195.↵
- 196.↵
- 197.↵
- 198.↵
- 199.↵
- 200.↵
- 201.↵
- 202.↵
- 203.↵
- 204.
- 205.↵
- 206.↵
- 207.↵
- 208.↵
- 209.↵
- 210.↵
- 211.↵
- 212.↵
- 213.↵
- 214.↵
- 215.↵
- 216.↵
- 217.↵
- 218.↵
- 219.↵
- 220.↵
- 221.↵





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Introduction
- Origins of Ventilator-Induced Lung Injury
- Current Concepts in Protective Ventilation Strategies for ARDS
- Respiratory Muscle Physiology and Mechanical Ventilation
- Recruitment and De-recruitment in ARDS
- VILI, Organ Cross Talk, ARDS Phenotypes, and Multi-Organ Dysfunction Syndrome
- “Wither Goest Thou?”: Integration of Ventilator and ICU Monitoring Data With Other Health Systems Data, ARDS Phenotypes, Computational Intelligence, and Closed-Loop Ventilation
- Footnotes
- References
- Figures & Data
- Info & Metrics
- References